T-Cell Lymphoblastic Lymphoma Arising in the Setting of Myeloid/Lymphoid Neoplasms with Eosinophilia
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Characterization of a Pediatric T-Cell Acute Lymphoblastic Leukemia Patient with Simultaneous LYL1 and LMO2 Rearrangements
View metadata, citation and similar papers at core.ac.uk brought to you by CORE provided by Erasmus University Digital Repository Articles and Brief Reports Acute Lymphoblastic Leukemia Characterization of a pediatric T-cell acute lymphoblastic leukemia patient with simultaneous LYL1 and LMO2 rearrangements Irene Homminga, 1 Maartje J. Vuerhard, 1 Anton W. Langerak, 2 Jessica Buijs-Gladdines, 1 Rob Pieters, 1 and Jules P.P. Meijerink 1 1Department of Pediatric Oncology/Hematology, Erasmus MC/Sophia Children’s Hospital, Rotterdam; and 2Department of Immunology, Erasmus MC, Rotterdam, The Netherlands ABSTRACT Translocation of the LYL1 oncogene are rare in T-cell acute consistently clustered along with cases having TAL1 or lymphoblastic leukemia, whereas the homologous TAL1 LMO2 rearrangements. Therefore, LYL1 -rearranged cases are gene is rearranged in approximately 20% of patients. not necessarily associated with immature T-cell develop - Previous gene-expression studies have identified an imma - ment, despite high LYL1 levels, but elicit a TALLMO expres - ture T-cell acute lymphoblastic leukemia subgroup with high sion signature. LYL1 expression in the absence of chromosomal aberrations. Molecular characterization of a t(7;19)(q34;p13) in a pediatric Key words: T-ALL, pediatric, LYL1, LMO2, rearrangements. T-cell acute lymphoblastic leukemia patient led to the identi - fication of a translocation between the TRB@ and LYL1 loci. Citation: Homminga I, Vuerhard MJ, Langerak AW, Buijs- Similar to incidental T-cell acute lymphoblastic leukemia Gladdines J, Pieters R, and Meijerink JPP. Characterization of a cases with synergistic, double translocations affecting pediatric T-cell acute lymphoblastic leukemia patient with simul - TAL1/2 and LMO1/2 oncogenes, this LYL1 -translocated taneous LYL1 and LMO2 rearrangements. -

FIP1L1 Gene FIP1 Like 1 (S
FIP1L1 gene FIP1 like 1 (S. cerevisiae) Normal Function The FIP1L1 gene provides instructions for making part of a protein complex named cleavage and polyadenylation specificity factor (CPSF). This complex of proteins plays an important role in processing molecules called messenger RNAs (mRNAs), which serve as the genetic blueprints for making proteins. The CPSF protein complex helps add a string of the RNA building block adenine to the mRNA, creating a polyadenine tail or poly(A) tail. The poly(A) tail is important for stability of the mRNA and for protein production from the blueprint. Health Conditions Related to Genetic Changes PDGFRA-associated chronic eosinophilic leukemia A deletion of genetic material from chromosome 4 brings together part of the FIP1L1 gene and part of another gene called PDGFRA, creating the FIP1L1-PDGFRA fusion gene. This mutation is a somatic mutation, which means it is acquired during a person's lifetime and is present only in certain cells. This fusion gene causes PDGFRA- associated chronic eosinophilic leukemia, which is a type of blood cell cancer characterized by an increased number of eosinophils, a type of white blood cell involved in allergic reactions. The FIP1L1-PDGFRA protein produced from the fusion gene has the function of the normal PDGFRA protein, which stimulates signaling pathways inside the cell that control many important cellular processes, such as cell growth and division (proliferation) and cell survival. Unlike the normal PDGFRA protein, however, the FIP1L1-PDGFRA protein is constantly turned on (constitutively activated), which means the cells are always receiving signals to proliferate. When the FIP1L1-PDGFRA fusion gene occurs in blood cell precursors, the growth of eosinophils (and occasionally other blood cells) is poorly controlled, leading to PDGFRA-associated chronic eosinophilic leukemia. -

R-Loop-Mediated Genome Instability in Mrna Cleavage and Polyadenylation Mutants
Downloaded from genesdev.cshlp.org on September 29, 2021 - Published by Cold Spring Harbor Laboratory Press R-loop-mediated genome instability in mRNA cleavage and polyadenylation mutants Peter C. Stirling,1,3 Yujia A. Chan,1,3 Sean W. Minaker,1,3 Maria J. Aristizabal,2 Irene Barrett,1 Payal Sipahimalani,1 Michael S. Kobor,2 and Philip Hieter1,4 1Michael Smith Laboratories, University of British Columbia, Vancouver, British Columbia V6T1Z4, Canada; 2Centre for Molecular Medicine and Therapeutics, Vancouver, British Columbia V5Z4H4, Canada Genome instability via RNA:DNA hybrid-mediated R loops has been observed in mutants involved in various aspects of transcription and RNA processing. The prevalence of this mechanism among essential chromosome instability (CIN) genes remains unclear. In a secondary screen for increased Rad52 foci in CIN mutants, representing ~25% of essential genes, we identified seven essential subunits of the mRNA cleavage and polyadenylation (mCP) machinery. Genome-wide analysis of fragile sites by chromatin immunoprecipitation (ChIP) and microarray (ChIP–chip) of phosphorylated H2A in these mutants supported a transcription-dependent mechanism of DNA damage characteristic of R loops. In parallel, we directly detected increased RNA:DNA hybrid formation in mCP mutants and demonstrated that CIN is suppressed by expression of the R-loop-degrading enzyme RNaseH. To investigate the conservation of CIN in mCP mutants, we focused on FIP1L1, the human ortholog of yeast FIP1, a conserved mCP component that is part of an oncogenic fusion in eosinophilic leukemia. We found that truncation fusions of yeast FIP1 analogous to those in cancer cause loss of function and that siRNA knockdown of FIP1L1 in human cells increases DNA damage and chromosome breakage. -

Hypereosinophilia in Granular Acute Bcell Lymphoblastic Leukemia With
bs_bs_banner Microscopic polyangiitis 543 and had been diagnosed with normocytic anemia.At that time, she References received treatment with an iron preparation because of a low 1 Ozen S, Ruperto N, Dillon MJ et al. EULAR/PReS endorsed con- serum iron level despite her normal ferritin level. We estimate that sensus criteria for the classification of childhood vasculitides. Ann. the patient already had mild alveolar hemorrhage at that time and Rheum. Dis. 2006; 65: 936–41. the bleeding later healed spontaneously. Among the cases of this 2 Vanoni F, Bettinelli A, Keller F, Bianchetti MG, Simonetti GD. disease reported from Japan, the disease tended to develop more Vasculitides associated with IgG antineutrophil cytoplasmic autoan- tibodies in childhood. Pediatr. Nephrol. 2010; 25: 205–12. frequently in the spring. In our patient, the disease developed twice 3 Kobayashi S, Inokuma S. Intrapulmonary hemorrhage in collagen- (both times in June), suggesting the possibility that infection vascular disease includes a spectrum of underlying conditions. triggered the disease onset. Although alveolar hemorrhage sub- Intern. Med. 2009; 48: 891–7. sided spontaneously in this case, lung fibrosis may develop in the 4 Hattori M, Kurayama H, Koitabashi Y. Antineutrophil cytoplasmic future if alveolar bleeding develops repeatedly and its diagnosis is autoantibody-associated glomerulonephritis in children. J. Am. Soc. Nephrol. 2001; 12: 1493–500. delayed. In the present case, treatment was started immediately 5 Peco-Antic A, Bonaci-Nikolic B, Basta-Jovanovic G et al. Child- after diagnosis and rapid aggravation of her renal function was hood microscopic polyangiitis associated with MPO-ANCA. prevented. When dealing with cases in whom unexplained nor- Pediatr. -

The Role of Noncoding Mutations in Blood Cancers Sunniyat Rahman and Marc R
© 2019. Published by The Company of Biologists Ltd | Disease Models & Mechanisms (2019) 12, dmm041988. doi:10.1242/dmm.041988 REVIEW The role of noncoding mutations in blood cancers Sunniyat Rahman and Marc R. Mansour* ABSTRACT survival in adult acute myeloid leukaemia (AML) and ALL are The search for oncogenic mutations in haematological malignancies still less than 50%, with significant treatment challenges including has largely focused on coding sequence variants. These variants treatment-resistant disease, clonal heterogeneity of the underlying have been critical in understanding these complex cancers in greater disease, treatment-associated toxicities and poor tolerance for detail, ultimately leading to better disease monitoring, subtyping and intensive treatment regimens, particularly in older patients with prognostication. In contrast, the search for oncogenic variants in the comorbidities (Kansagra et al., 2018). Because of this, there is a noncoding genome has proven to be challenging given the vastness significant impetus to discover new genetic aberrations, including of the search space, the intrinsic difficulty in assessing the impact of driver mutations (see Box 1 for a glossary of terms), that may variants that do not code for functional proteins, and our still primitive provide insight into the mechanisms of malignant haematopoiesis, understanding of the function harboured by large parts of the as well as offering novel therapeutic opportunities. noncoding genome. Recent studies have broken ground on this Detailed genetic characterisation of haematological malignancies quest, identifying somatically acquired and recurrent mutations in the has already identified alterations that are now being used for better noncoding genome that activate the expression of proto-oncogenes. diagnosis, prognostication, subtype identification and to inform In this Review, we explore some of the best-characterised examples therapeutic decisions (Taylor et al., 2017). -

1 Supporting Information for a Microrna Network Regulates
Supporting Information for A microRNA Network Regulates Expression and Biosynthesis of CFTR and CFTR-ΔF508 Shyam Ramachandrana,b, Philip H. Karpc, Peng Jiangc, Lynda S. Ostedgaardc, Amy E. Walza, John T. Fishere, Shaf Keshavjeeh, Kim A. Lennoxi, Ashley M. Jacobii, Scott D. Rosei, Mark A. Behlkei, Michael J. Welshb,c,d,g, Yi Xingb,c,f, Paul B. McCray Jr.a,b,c Author Affiliations: Department of Pediatricsa, Interdisciplinary Program in Geneticsb, Departments of Internal Medicinec, Molecular Physiology and Biophysicsd, Anatomy and Cell Biologye, Biomedical Engineeringf, Howard Hughes Medical Instituteg, Carver College of Medicine, University of Iowa, Iowa City, IA-52242 Division of Thoracic Surgeryh, Toronto General Hospital, University Health Network, University of Toronto, Toronto, Canada-M5G 2C4 Integrated DNA Technologiesi, Coralville, IA-52241 To whom correspondence should be addressed: Email: [email protected] (M.J.W.); yi- [email protected] (Y.X.); Email: [email protected] (P.B.M.) This PDF file includes: Materials and Methods References Fig. S1. miR-138 regulates SIN3A in a dose-dependent and site-specific manner. Fig. S2. miR-138 regulates endogenous SIN3A protein expression. Fig. S3. miR-138 regulates endogenous CFTR protein expression in Calu-3 cells. Fig. S4. miR-138 regulates endogenous CFTR protein expression in primary human airway epithelia. Fig. S5. miR-138 regulates CFTR expression in HeLa cells. Fig. S6. miR-138 regulates CFTR expression in HEK293T cells. Fig. S7. HeLa cells exhibit CFTR channel activity. Fig. S8. miR-138 improves CFTR processing. Fig. S9. miR-138 improves CFTR-ΔF508 processing. Fig. S10. SIN3A inhibition yields partial rescue of Cl- transport in CF epithelia. -

PRODUCT SPECIFICATION Prest Antigen FIP1L1 Product Datasheet
PrEST Antigen FIP1L1 Product Datasheet PrEST Antigen PRODUCT SPECIFICATION Product Name PrEST Antigen FIP1L1 Product Number APrEST79770 Gene Description factor interacting with PAPOLA and CPSF1 Alternative Gene DKFZp586K0717 Names Corresponding Anti-FIP1L1 (HPA037475) Antibodies Description Recombinant protein fragment of Human FIP1L1 Amino Acid Sequence Recombinant Protein Epitope Signature Tag (PrEST) antigen sequence: KANSSVGKWQDRYGRAESPDLRRLPGAIDVIGQTITISRVEGRRRANENS NIQVLSERSATEVDNNFSKPPPFFPPGAPPT Fusion Tag N-terminal His6ABP (ABP = Albumin Binding Protein derived from Streptococcal Protein G) Expression Host E. coli Purification IMAC purification Predicted MW 27 kDa including tags Usage Suitable as control in WB and preadsorption assays using indicated corresponding antibodies. Purity >80% by SDS-PAGE and Coomassie blue staining Buffer PBS and 1M Urea, pH 7.4. Unit Size 100 µl Concentration Lot dependent Storage Upon delivery store at -20°C. Avoid repeated freeze/thaw cycles. Notes Gently mix before use. Optimal concentrations and conditions for each application should be determined by the user. Product of Sweden. For research use only. Not intended for pharmaceutical development, diagnostic, therapeutic or any in vivo use. No products from Atlas Antibodies may be resold, modified for resale or used to manufacture commercial products without prior written approval from Atlas Antibodies AB. Warranty: The products supplied by Atlas Antibodies are warranted to meet stated product specifications and to conform to label descriptions when used and stored properly. Unless otherwise stated, this warranty is limited to one year from date of sales for products used, handled and stored according to Atlas Antibodies AB's instructions. Atlas Antibodies AB's sole liability is limited to replacement of the product or refund of the purchase price. -

Land Urbanization in Central Italy: 50 Years of Evolution
This article was downloaded by: [Bernardino Romano] On: 22 July 2014, At: 10:32 Publisher: Taylor & Francis Informa Ltd Registered in England and Wales Registered Number: 1072954 Registered office: Mortimer House, 37-41 Mortimer Street, London W1T 3JH, UK Journal of Land Use Science Publication details, including instructions for authors and subscription information: http://www.tandfonline.com/loi/tlus20 Land urbanization in Central Italy: 50 years of evolution Bernardino Romanoa & Francesco Zulloa a Department of Civil Engineering, Architecture and Environment (DICEAA), University of L’Aquila, Campo di Pile, L’Aquila, Italy Accepted author version posted online: 04 Dec 2012.Published online: 06 Feb 2013. To cite this article: Bernardino Romano & Francesco Zullo (2014) Land urbanization in Central Italy: 50 years of evolution, Journal of Land Use Science, 9:2, 143-164, DOI: 10.1080/1747423X.2012.754963 To link to this article: http://dx.doi.org/10.1080/1747423X.2012.754963 PLEASE SCROLL DOWN FOR ARTICLE Taylor & Francis makes every effort to ensure the accuracy of all the information (the “Content”) contained in the publications on our platform. However, Taylor & Francis, our agents, and our licensors make no representations or warranties whatsoever as to the accuracy, completeness, or suitability for any purpose of the Content. Any opinions and views expressed in this publication are the opinions and views of the authors, and are not the views of or endorsed by Taylor & Francis. The accuracy of the Content should not be relied upon and should be independently verified with primary sources of information. Taylor and Francis shall not be liable for any losses, actions, claims, proceedings, demands, costs, expenses, damages, and other liabilities whatsoever or howsoever caused arising directly or indirectly in connection with, in relation to or arising out of the use of the Content. -

LMO T-Cell Translocation Oncogenes Typify Genes Activated by Chromosomal Translocations That Alter Transcription and Developmental Processes
Downloaded from genesdev.cshlp.org on September 25, 2021 - Published by Cold Spring Harbor Laboratory Press PERSPECTIVE LMO T-cell translocation oncogenes typify genes activated by chromosomal translocations that alter transcription and developmental processes Terence H. Rabbitts1 Medical Research Council (MRC) Laboratory of Molecular Biology, Division of Protein and Nucleic Acid Chemistry, Cambridge CB2 2QH, UK The cytogenetic analysis of tumors, particularly those of ment (immunoglobulin or T-cell receptor) occurs and oc- hematopoietic origin, has revealed that reciprocal chro- casionally mediates chromosomal translocation. This mosomal translocations are recurring features of these type of translocation causes oncogene activation result- tumors. Further from the initial recognition of the trans- ing from the new chromosomal environment of the re- location t(9;22) (Nowell and Hungerford 1960; Rowley arranged gene. In general, this means inappropriate gene 1973), it has become clear that particular chromosomal expression. The second, and probably the most common translocations are found consistently in specific tumor outcome of chromosomal translocations, is gene fusion subtypes. A principle example of this is the translocation in which exons from a gene on each of the involved chro- t(8;14)(q24;q32.1) invariably found in the human B cell mosomes are linked after the chromosomal transloca- tumor Burkitt’s lymphoma (Manolov and Manolov 1972; tion, resulting in a fusion mRNA and protein. This type Zech et al. 1976). The link with the immunoglobulin of event is found in many cases of hematopoietic tumors H-chain locus on chromosome 14, band q32.1 (Croce et and in the sarcomas. al. 1979; Hobart et al. -

California Studies in Food and Culture Darra Goldstein, Editor
California Studies in Food and Culture Darra Goldstein, Editor Th e publisher gratefully acknowledges the generous support of the Ahmanson Foundation Humanities Endowment Fund of the University of California Press Foundation. Th e publisher also gratefully acknowledges the generous support of the Humanities Endowment Fund of the University of California Press Foundation. Popes, Peasants, and Shepherds Popes, Peasants, and Shepherds recipes and lore from rome and lazio Oretta Zanini De Vita Translated by Maureen B. Fant With a foreword by Ernesto Di Renzo University of California Press berkeley los angeles london Series page: Caulifl ower grower selling his harvest in the streets of Rome (Biblioteca Clementina, Anzio) Frontispiece: Bartolomeo Pinelli, Temple of the Sibyl at Tivoli (Biblioteca Clementina, Anzio) University of California Press, one of the most distinguished university presses in the United States, enriches lives around the world by advancing scholarship in the humanities, social sciences, and natural sciences. Its activities are sup- ported by the UC Press Foundation and by philanthropic contributions from individuals and institutions. For more information, visit www .ucpress .edu . University of California Press Berkeley and Los Angeles, California University of California Press, Ltd. London, En gland © 2013 by Oretta Zanini De Vita A revised and expanded edition of Il Lazio a tavola: Guida gastronomica tra storia e tradizioni, originally published in Italian and simultaneously in English as Th e Food of Rome and Lazio: History, Folklore, and Recipes. Library of Congress Cataloging- in- Publication Data Zanini De Vita, Oretta, 1936– [Lazio a tavola. En glish] Popes, peasants, and shepherds : recipes and lore from Rome and Lazio / Oretta Zanini De Vita ; Translated by Maureen B. -
Letters to the Editor
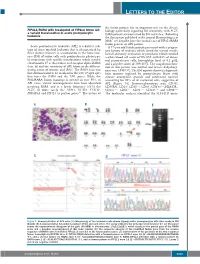
LETTERS TO THE EDITOR the fusion partner has an important role on the disease FIP1L1/RARA with breakpoint at FIP1L1 intron 13: biology particularly regarding RA sensitivity, with PLZF- a variant translocation in acute promyelocytic 4 leukemia RARA patients characterized by RA resistance. Following the description published in the journal Haematologica in 2008, 5 we describe here the second case of FIP1L1/RARA fusion gene in an APL patient. Acute promyelocytic leukemia (APL) is a distinct sub - A 77-year old female patient presented with a progres - type of acute myeloid leukemia that is characterized by sive history of asthenia which lasted for several weeks. three distinct features: i) accumulation in the bone mar - Initial laboratory evaluation of peripheral blood revealed row (BM) of tumor cells with promyelocytic phenotype; a white blood cell count of 59 ¥10 9/L with 84% of abnor - ii) association with specific translocations which involve mal promyelocyte cells, hemoglobin level of 9.2 g/dL, chromosome 17 at the retinoic acid receptor alpha ( RARA ) and a platelet count of 109 ¥10 9/L. The coagulation func - locus ; iii) and the sensitivity of APL blasts to the differen - tion in this patient was normal and lactate dehydroge - 1 tiating action of retinoic acid (RA). The RARA locus was nase was 1,938 U/L. The BM aspirate showed a hypercel - first demonstrated to be involved in the t(15;17)(q22;q21) lular marrow replaced by promyelocyte blasts with that fuses the RARA and the PML genes. While the intense azurophilic granule and prominent nucleoli PML/RARA fusion transcript is present in over 95% of accounting for 93% of all nucleated cells, suggestive of APL cases, variant rearrangements have been identified APL (Figure 1A). -

English Speaking Doctors and Medical Facilities in the Rome Consular District
English Speaking Doctors and Medical Facilities in the Rome Consular District (The Rome district contains the regions of Lazio, Abruzzo, Marche, Umbria and Sardinia) Disclaimer: The U.S. Embassy in Rome assumes no responsibility for the professional ability or reputation of the persons or medical facilities whose names appear on the lists. Inclusion on these lists is in no way an endorsement by the Department of State or the U.S. Embassy. Names are listed alphabetically and the order in which they appear has no other significance. The information on the lists regarding professional credentials and areas of expertise is provided directly by the medical professional, medical facility or ambulance service. The U.S. Embassy is not in a position to vouch for such information. You may receive additional information about the individuals by contacting the local medical boards and associations or the local licensing authorities. • Public Hospitals in Rome • Private Hospitals in Rome • List of Doctors in Rome • Other Public Hospitals within the Consular District of Rome • Ambulance services in Rome • Laboratories in Rome • Pharmacies & Opticians in Rome PUBLIC HOSPITALS IN ROME PLEASE NOTE: The following color code has been created to facilitate locating public hospitals in the city of Rome. COLOR CODE: Black = Center (for example, Vatican, U.S. Embassy, Piazza del Popolo, Colosseum, Pantheon, Termini Train Station), Blue = West (for example, Fiumicino Airport, Via Aurelia, San Giovanni Cathedral, Villa Pamphili), Red = North (for example, Viale