Pharmacological Targeting of the Atherogenic Dyslipidemia Complex: the Next Frontier in CVD Prevention Beyond Lowering LDL Cholesterol
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Cholesteryl Ester-Transfer Protein Inhibitors Stimulate Aldosterone Biosynthesis in Adipocytes Through Nox-Dependent Processes S
Supplemental material to this article can be found at: http://jpet.aspetjournals.org/content/suppl/2015/01/23/jpet.114.221002.DC1 1521-0103/353/1/27–34$25.00 http://dx.doi.org/10.1124/jpet.114.221002 THE JOURNAL OF PHARMACOLOGY AND EXPERIMENTAL THERAPEUTICS J Pharmacol Exp Ther 353:27–34, April 2015 Copyright ª 2015 by The American Society for Pharmacology and Experimental Therapeutics Cholesteryl Ester-Transfer Protein Inhibitors Stimulate Aldosterone Biosynthesis in Adipocytes through Nox-Dependent Processes s Francisco J. Rios, Karla B. Neves, Aurelie Nguyen Dinh Cat, Sarah Even, Roberto Palacios, Augusto C. Montezano, and Rhian M. Touyz Institute of Cardiovascular and Medical Sciences, British Heart Foundation Glasgow Cardiovascular Research Centre, University of Glasgow, Glasgow, Scotland, United Kingdom (F.J.R., A.N.D.C., S.E., A.C.M., R.M.T.); Faculty of Pharmaceutical Sciences of Ribeirao Preto, University of Sao Paulo, Ribeirao Preto, Brazil (K.B.N.); and Departamento de Bioquímica, Fisiología y Genética Molecular Facultad de CC. de la Salud, Universidad Rey Juan Carlos, Madrid, Spain (R.P.) Downloaded from Received October 29, 2014; accepted January 22, 2015 ABSTRACT Hyperaldosteronism and hypertension were unexpected side whereby CETP inhibitors mediate effects, cells were pretreated effects observed in trials of torcetrapib, a cholesteryl ester-transfer with inhibitors of Nox1/Nox4 [GKT137831; 2-(2-chlorophenyl)-4-[3- jpet.aspetjournals.org protein (CETP) inhibitor that increases high-density lipoprotein. (dimethylamino)phenyl]-5-methyl-1H-pyrazolo[4,3-c]pyridine- Given that CETP inhibitors are lipid soluble, accumulate in adipose 3,6(2H,5H)-dione], Nox1 (ML171 [2-acetylphenothiazine]), mitochondria tissue, and have binding sites for proteins involved in adipogenesis, (rotenone), and STAT3 (S3I-201 [2-hydroxy-4-(((4-methylphenyl) and that adipocytes are a source of aldosterone, we questioned sulfonyloxy)acetyl)amino)-benzoic acid]). -

Next-Generation Sequencing Transforms Today's Biology
Evolving new therapies for the prevention of atherosclerosis: a glimpse of the near future G.K. Hovingh MD PhD ([email protected]) Department of Vascular Medicine AMC Amsterdam The Netherlands Today BMJ 2013;347:f544 www.chinadaily.com.cn/life/2009- 04/21/content_7698500.htm What we know - CVD major burden - LDL-C causally related with CVD - LDL-C goals: the lower the better - Statins : corner stone in therapy How well do we do? Reduction in MACE statin vs placebo (%) 0 -30 Potential for further risk reduction -100 Where do we go? Reduction in MACE statin vs placebo (%) 0 Potential for further -30 risk reduction -50 = further LDL-C lowering? and or Additional Rx? -100 A glance at the future… Atherosclerosis • LDL • HDL • TG • Lp(a) • Inflammation Lipid Modifying Drugs • Cholesterol absorption inhibitors • Squalene synthase inhibitors (SSI) • Microsomal triglyceride transfer protein (MTP) inhibitors • Acyl coenzyme A acyltransferase (ACAT) inhibitors • Diacylglycerol acyltransferase (DGAT) inhibitors • Thyroxin receptor agonists • ApoB mRNA antisense drugs • PCSK9 antibodies • ApoA1-based strategies (iv) • Cholesterol ester transfer protein (CETP) inhibitors ApoA-1 based therapy ApoA1 Mimetics, such as APL-180 Novartis Full-length ApoA1, such as ApoA1 Cerenis Therapeutics Pre-Beta HDL, as generated by delipidation, HDL Therapeutics Inc. Reconstituted HDL, CSL Ltd. ApoA1 Milano MDCO216, The Medicines Company Trimeric ApoA1, Borean Pharma and now Roche RVX-208, as developed by Resverlogix Fx-5A, as developed by Kinemed Inc. HDL -

Studies on the Absorptive Defect for Triglyceride in Abetalipoproteinemia * P
Jownal of Clinical Investigation Vol. 46, No. 1, 1967 Studies on the Absorptive Defect for Triglyceride in Abetalipoproteinemia * P. 0. WAYS,t C. M. PARMENTIER, H. J. KAYDEN,4 J. W. JONES,§ D. R. SAUNDERS,|| AND C. E. RUBIN 1J (From the Department of Medicine, University of Washington School of Medicine, Seattle, Wash., and the Department of Medicine, New York University School of Medicine, New York, N. Y.) Summary. The nature of the gastrointestinal absorptive defect for triglycer- ide in three subjects with abetalipoproteinemia has been investigated by studying peroral biopsies of the gastrointestinal mucosa. The following con- clusions were reached. 1) In confirmation of other studies, the abnormal vacuoles within the du- odenal absorptive cells of these individuals were lipophilic. 2) On chemical analysis there was significantly more mucosal lipid than found in normal fasting specimens, and almost the entire increase was due to triglyceride. 3) This excess mucosal lipid was reduced by a low fat diet, but even after 34 days on such a diet there was still an excess of lipophilic material near the villus tip and increased quantities of total lipid and triglyceride when com- pared with material from normal subjects similarly treated. 4) Although there are demonstrable qualitative changes in mucosal and plasma lipids after an acute fat load, they are not quantitatively as great as in normal individuals. Fat balance studies and the qualitative changes in plasma and tissue lipids that do occur after more extended periods on different types of dietary fat do indicate that a considerable percentage of the dietary fat is assimilated. -
Statins and Lipid Lowering Agents
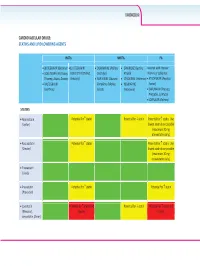
CARDIOVASCULAR Z/Ks^h>ZZh'^͗ ^dd/E^E>/W/>KtZ/E''Ed^ /E^d/Ɛ EEZd/Ɛ W/Ɛ x/d'Zs/Z ;ŝŬƚĂƌǀLJͿ x>s/d'Zs/Zͬ x KZs/Z/E ;WŝĨĞůƚƌŽ͕ x &s/ZE ;^ƵƐƚŝǀĂ͕ ŽŽƐƚĞĚǁŝƚŚƌŝƚŽŶĂǀŝƌ xK>hd'Zs/Z ;dŝǀŝĐĂLJ͕ K//^dd ;^ƚƌŝďŝůĚ͕ ĞůƐƚƌŝŐŽͿ ƚƌŝƉůĂͿ ;EŽƌǀŝƌͿŽƌĐŽďŝĐŝƐƚĂƚ dƌŝƵŵĞƋ͕:ƵůƵĐĂ͕ŽǀĂƚŽͿ 'ĞŶǀŽLJĂͿ x Z/>W/s/Z/E ;ĚƵƌĂŶƚ͕ x dZs/Z/E ;/ŶƚĞůĞŶĐĞͿ xdEs/Z ;ZĞLJĂƚĂnj͕ xZ>d'Zs/Z ŽŵƉůĞƌĂ͕KĚĞĨƐĞLJ͕ x Es/ZW/E ǀŽƚĂnjͿ ;/ƐĞŶƚƌĞƐƐͿ :ƵůƵĐĂͿ ;sŝƌĂŵƵŶĞͿ xZhEs/Z ;WƌĞnjŝƐƚĂ͕ WƌĞnjĐŽďŝdž͕^LJŵƚƵnjĂͿ x>KW/Es/Z ;<ĂůĞƚƌĂͿ ^dd/E^ xƚŽƌǀĂƐƚĂƚŝŶ WŽƚĞŶƚŝĂůĨŽƌ nƐƚĂƚŝŶ WŽƚĞŶƚŝĂůĨŽƌ pƐƚĂƚŝŶ WŽƚĞŶƚŝĂůĨŽƌ nƐƚĂƚŝŶ͘hƐĞ ;>ŝƉŝƚŽƌͿ ůŽǁĞƐƚƐƚĂƚŝŶĚŽƐĞƉŽƐƐŝďůĞ ;ŵĂdžŝŵƵŵϮϬŵŐ ĂƚŽƌǀĂƐƚĂƚŝŶĚĂŝůLJͿ͘ xZŽƐƵǀĂƐƚĂƚŝŶ WŽƚĞŶƚŝĂůĨŽƌ nƐƚĂƚŝŶ WŽƚĞŶƚŝĂůĨŽƌ nƐƚĂƚŝŶ͘hƐĞ ;ƌĞƐƚŽƌͿ ůŽǁĞƐƚƐƚĂƚŝŶĚŽƐĞƉŽƐƐŝďůĞ ;ŵĂdžŝŵƵŵϭϬŵŐ ƌŽƐƵǀĂƐƚĂƚŝŶĚĂŝůLJͿ͘ xWŝƚĂǀĂƐƚĂƚŝŶ ;>ŝǀĂůŽͿ xWƌĂǀĂƐƚĂƚŝŶ WŽƚĞŶƚŝĂůĨŽƌ nƐƚĂƚŝŶ WŽƚĞŶƚŝĂůĨŽƌ nƐƚĂƚŝŶ ;WƌĂǀĂĐŚŽůͿ x>ŽǀĂƐƚĂƚŝŶ WŽƚĞŶƚŝĂůĨŽƌ nƐƚĂƚŝŶĂŶĚ WŽƚĞŶƚŝĂůĨŽƌ pƐƚĂƚŝŶ WŽƚĞŶƚŝĂůĨŽƌ nƐƚĂƚŝŶĂŶĚ ;DĞǀĂĐŽƌͿ͕ ƚŽdžŝĐŝƚLJ ƚŽdžŝĐŝƚLJ ƐŝŵǀĂƐƚĂƚŝŶ ;ŽĐŽƌͿ CARDIOVASCULAR INSTIs NNRTIs PIs xBICTEGRAVIR (Biktarvy) xELVITEGRAVIR/ x DORAVIRINE (Pifeltro, x EFAVIRENZ (Sustiva, Boosted with ritonavir xDOLUTEGRAVIR (Tivicay, COBICISTAT (Stribild, Delstrigo) Atripla) (Norvir) or cobicistat Triumeq, Juluca) Genvoya) x RILPIVIRINE (Edurant, x ETRAVIRINE (Intelence) xATAZANAVIR (Reyataz, xRALTEGRAVIR Complera, Odefsey, x NEVIRAPINE Evotaz) (Isentress) Juluca) (Viramune) xDARUNAVIR (Prezista, Prezcobix, Symtuza) xLOPINAVIR (Kaletra) FIBRATES xFenofibrate, bezafibrate, gemfibrozil CHOLESTEROL ABSORPTION INHIBITOR xEzetimibe -

Genetic Testing for Familial Hypercholesterolemia AHS – M2137
Corporate Medical Policy Genetic Testing for Familial Hypercholesterolemia AHS – M2137 File Name: genetic_testing_for_familial_hypercholesterolemia Origination: 01/01/2019 Last CAP Review: 07/2021 Next CAP Review: 07/2022 Last Review: 07/2021 Description of Procedure or Service Definitions Familial hypercholesterolemia (FH) is a genetic condition that results in premature atherosclerotic cardiovascular disease due to lifelong exposure to elevated low-density lipoprotein cholesterol (LDL-C) levels (Sturm et al., 2018). FH encompasses multiple clinical phenotypes associated with distinct molecular etiologies. The most common is an autosomal dominant disorder caused by mutations in one of three genes, low-density lipoprotein receptor (LDLR), apolipoprotein B-100 (APOB), and proprotein convertase subtilisin-like kexin type 9 (PCSK9) (Ahmad et al., 2016; Goldberg et al., 2011). Rare autosomal recessive disease results from mutation in low-density lipoprotein receptor adaptor protein (LDLRAP) (Garcia et al., 2001). Related Policies Cardiovascular Disease Risk Assessment AHS – G2050 ***Note: This Medical Policy is complex and technical. For questions concerning the technical language and/or specific clinical indications for its use, please consult your physician. Policy BCBSNC will provide coverage for genetic testing for familial hypercholesterolemia when it is determined the medical criteria or reimbursement guidelines below are met. Benefits Application This medical policy relates only to the services or supplies described herein. Please refer to the Member's Benefit Booklet for availability of benefits. Member's benefits may vary according to benefit design; therefore member benefit language should be reviewed before applying the terms of this medical policy. When Genetic Testing for Familial Hypercholesterolemia is covered 1. Genetic testing to establish a molecular diagnosis of Familial Hypercholesterolemia is considered medically necessary when BOTH of the following criteria are met: A. -

Bovine Milk Lipoprotein Lipase Transfers Tocopherol to Human Fibroblasts During Triglyceride Hydrolysis in Vitro Maret G
Bovine Milk Lipoprotein Lipase Transfers Tocopherol to Human Fibroblasts during Triglyceride Hydrolysis In Vitro Maret G. Traber, Thomas Olivecrona, and Herbert J. Kayden Department ofMedicine, New York University School ofMedicine, New York 10016; University of Umea, Umea, Sweden Abstract vitamin E, have minimal or absent neurological abnormalities, but those who have not been supplemented have a progressive Lipoprotein lipase appears to function as the mechanism by which dietary vitamin E (tocopherol) is transferred from chy- degeneration of the peripheral nervous system resulting in ataxia and characteristic pathologic changes in the central lomicrons to tissues. In patients with lipoprotein lipase defi- nervous system (5). These neuropathologic changes have been more than 85% of both the and ciency, circulating triglyceride observed in patients with cholestatic liver disease in whom the tocopherol is contained in the chylomicron fraction. The studies in low to absent levels of bile salts in the intestine results in an presented here show that the vitro addition of bovine milk inability to absorb vitamin E (3). Oral supplementation with to in the of lipoprotein lipase (lipase) chylomicrons presence pharmacologic amounts of the vitamin (6), or or fibroblasts bovine albumin pagenteral human erythrocytes (and serum administration of the vitamin (when there is a complete resulted in the hydrolysis of triglyceride and the [BSAJ) the absence of intestinal bile) (7) results in the prevention of transfer of both acids and to the in the fatty tocopherol cells; further of the nervous system. The studies in absence of no in cellular was deterioration lipase, increase tocopherol these two groups ofpatients have demonstrated the importance The incubation was to include detectable. -

PHARMACEUTICAL APPENDIX to the TARIFF SCHEDULE 2 Table 1
Harmonized Tariff Schedule of the United States (2020) Revision 19 Annotated for Statistical Reporting Purposes PHARMACEUTICAL APPENDIX TO THE HARMONIZED TARIFF SCHEDULE Harmonized Tariff Schedule of the United States (2020) Revision 19 Annotated for Statistical Reporting Purposes PHARMACEUTICAL APPENDIX TO THE TARIFF SCHEDULE 2 Table 1. This table enumerates products described by International Non-proprietary Names INN which shall be entered free of duty under general note 13 to the tariff schedule. The Chemical Abstracts Service CAS registry numbers also set forth in this table are included to assist in the identification of the products concerned. For purposes of the tariff schedule, any references to a product enumerated in this table includes such product by whatever name known. -

Lipodystrophy Due to Adipose Tissue Specific Insulin Receptor
Page 1 of 50 Diabetes Lipodystrophy Due to Adipose Tissue Specific Insulin Receptor Knockout Results in Progressive NAFLD Samir Softic1,2,#, Jeremie Boucher1,3,#, Marie H. Solheim1,4, Shiho Fujisaka1, Max-Felix Haering1, Erica P. Homan1, Jonathon Winnay1, Antonio R. Perez-Atayde5, and C. Ronald Kahn1. 1 Section on Integrative Physiology and Metabolism, Joslin Diabetes Center and Department of Medicine, Harvard Medical School, Boston, MA 2 Division of Gastroenterology, Hepatology and Nutrition, Boston Children’s Hospital, Boston, MA 3 Cardiovascular and Metabolic Diseases iMed, AstraZeneca R&D, 431 83 Mölndal, Sweden (current address) 4 KG Jebsen Center for Diabetes Research, Department of Clinical Science, University of Bergen, Bergen, Norway 5 Department of Pathology, Boston Children’s Hospital, and Harvard Medical School, Boston, MA # These authors contributed equally to this work. Corresponding author: C. Ronald Kahn, MD Joslin Diabetes Center One Joslin Place Boston, MA 02215 Phone: (617)732-2635 Fax:(617)732-2487 E-mail: [email protected] Keywords: Insulin receptors, IGF-1 receptors, lipodystrophy, diabetes, dyslipidemia, fatty liver, liver tumor, NAFLD, NASH. Running title: Lipodystrophic mice develop progressive NAFLD 1 Diabetes Publish Ahead of Print, published online May 10, 2016 Diabetes Page 2 of 50 SUMMARY Ectopic lipid accumulation in the liver is an almost universal feature of human and rodent models of generalized lipodystrophy and also is a common feature of type 2 diabetes, obesity and metabolic syndrome. Here we explore the progression of fatty liver disease using a mouse model of lipodystrophy created by a fat-specific knockout of the insulin receptor (F-IRKO) or both IR and insulin-like growth factor-1 receptor (F- IR/IGF1RKO). -

Clinofibrate Improved Canine Lipid Metabolism in Some but Not All Breeds
NOTE Internal Medicine Clinofibrate improved canine lipid metabolism in some but not all breeds Yohtaro SATO1), Nobuaki ARAI2), Hidemi YASUDA3) and Yasushi MIZOGUCHI4)* 1)Graduate School of Agriculture, Meiji University, 1-1-1 Higashimita, Tama-ku, Kawasaki, Kanagawa 214-8571, Japan 2)Spectrum Lab Japan, 1-5-22-201 Midorigaoka, Meguro-ku, Tokyo 152-0034, Japan 3)Yasuda Veterinary Clinic, 1-5-22 Midorigaoka, Meguro-ku, Tokyo 152-0034, Japan 4)School of Agriculture, Meiji University, 1-1-1 Higashimita, Tama-ku, Kawasaki, Kanagawa 214-8571, Japan ABSTRACT. The objectives of this study were to assess if Clinofibrate (CF) treatment improved J. Vet. Med. Sci. lipid metabolism in dogs, and to clarify whether its efficacy is influenced by canine characteristics. 80(6): 945–949, 2018 We collected medical records of 306 dogs and performed epidemiological analyses. Lipid values of all lipoproteins were significantly decreased by CF medication, especially VLDL triglyceride doi: 10.1292/jvms.17-0703 (TG) concentration (mean reduction rate=54.82%). However, 17.65% of dogs showed drug refractoriness in relation to TG level, and Toy Poodles had a lower CF response than other breeds (OR=5.36, 95% CI=2.07–13.90). Therefore, our study suggests that genetic factors may have an Received: 22 December 2017 effect on CF response, so genetic studies on lipid metabolism-related genes might be conducted Accepted: 9 March 2018 to identify variations in CF efficacy. Published online in J-STAGE: KEY WORDS: clinofibrate, descriptive epidemiology, drug response, dyslipidemia, Toy Poodle 26 March 2018 High serum cholesterol (Cho) and triglyceride (TG) concentrations in dogs are caused by various factors such as lack of exercise, high fat diets, obesity, neutralization, age, diseases and breed [6, 21, 24]. -

Dysfunctional High-Density Lipoproteins in Type 2 Diabetes Mellitus: Molecular Mechanisms and Therapeutic Implications
Journal of Clinical Medicine Review Dysfunctional High-Density Lipoproteins in Type 2 Diabetes Mellitus: Molecular Mechanisms and Therapeutic Implications Isabella Bonilha 1 , Francesca Zimetti 2,* , Ilaria Zanotti 2 , Bianca Papotti 2 and Andrei C. Sposito 1,* 1 Atherosclerosis and Vascular Biology Laboratory (AtheroLab), Cardiology Department, State University of Campinas (Unicamp), Campinas 13084-971, Brazil; [email protected] 2 Department of Food and Drug, University of Parma, 43124 Parma, Italy; [email protected] (I.Z.); [email protected] (B.P.) * Correspondence: [email protected] (F.Z.); [email protected] (A.C.S.); Tel.: +39-05-2190-6172 (F.Z.); +55-19-3521-7098 (A.C.S.); Fax: +55-1-9328-9410 (A.C.S.) Abstract: High density lipoproteins (HDLs) are commonly known for their anti-atherogenic prop- erties that include functions such as the promotion of cholesterol efflux and reverse cholesterol transport, as well as antioxidant and anti-inflammatory activities. However, because of some chronic inflammatory diseases, such as type 2 diabetes mellitus (T2DM), significant changes occur in HDLs in terms of both structure and composition. These alterations lead to the loss of HDLs’ physiological functions, to transformation into dysfunctional lipoproteins, and to increased risk of cardiovascular disease (CVD). In this review, we describe the main HDL structural/functional alterations observed in T2DM and the molecular mechanisms involved in these T2DM-derived modifications. Finally, the main available therapeutic interventions targeting HDL in diabetes are discussed. Citation: Bonilha, I.; Zimetti, F.; Keywords: high density lipoprotein; type 2 diabetes mellitus; HDL function; glycation; oxidation; Zanotti, I.; Papotti, B.; Sposito, A.C. -

Effects of Clofibrate Derivatives on Hyperlipidemia Induced by a Cholesterol-Free, High-Fructose Diet in Rats
Showa Univ. J. Med. Sci. 7(2), 173•`182, December 1995 Original Effects of Clofibrate Derivatives on Hyperlipidemia Induced by a Cholesterol-Free, High-Fructose Diet in Rats Hideyukl KURISHIMA,Sadao NAKAYAMA,Minoru FURUYA and Katsuji OGUCHI Abstract: The effects of the clofibrate derivatives fenofibrate (FF), bezafibrate (BF), and clinofibrate (CF), on hyperlipidemia induced by a cholesterol-free, high-fructose diet (HFD) in rats were investigated. Feeding of HFD for 2 weeks increased the high-density lipoprotein subfraction (HDL1) and decreased the low-density lipoprotein (LDL) fraction. The levels of total cholesterol (TC), free cholesterol, triglyceride (TG), and phospholipid in serum were increased by HFD feeding. Administration of CF inhibited the increase in HDL1 content. All three agents inhibited the decrease in LDL level. Both BF and CF decreased VLDL level. Administration of FF, BF, or CF inhibited the increases of serum lipids, especially that of TC and TG. The inhibitory effects of CF on HFD- induced increases in HDL1, TC, and TG were greater than those of FF and BF. These results demonstrate that FF, BF, and CF improve the intrinsic hyper- lipidemia induced by HFD feeding in rats. Key words: fenofibrate, bezafibrate, clinofibrate, fructose-induced hyperlipide- mia, lipoprotein. Introduction Clofibrate is one of the most effective antihypertriglycedemic agents currently available. However, because of its adverse effects, such as hepatomegaly1, several derivatives, such as clinofibrate (CF) and bezafibrate (BF) have been developed which are more effective and have fewer adverse effects. For example, it has been shown that the hypolipidemic effect of CF is greater than that of clofibrate while its tendency to produce hepatomegaly is less1. -

Partial Agreement in the Social and Public Health Field
COUNCIL OF EUROPE COMMITTEE OF MINISTERS (PARTIAL AGREEMENT IN THE SOCIAL AND PUBLIC HEALTH FIELD) RESOLUTION AP (88) 2 ON THE CLASSIFICATION OF MEDICINES WHICH ARE OBTAINABLE ONLY ON MEDICAL PRESCRIPTION (Adopted by the Committee of Ministers on 22 September 1988 at the 419th meeting of the Ministers' Deputies, and superseding Resolution AP (82) 2) AND APPENDIX I Alphabetical list of medicines adopted by the Public Health Committee (Partial Agreement) updated to 1 July 1988 APPENDIX II Pharmaco-therapeutic classification of medicines appearing in the alphabetical list in Appendix I updated to 1 July 1988 RESOLUTION AP (88) 2 ON THE CLASSIFICATION OF MEDICINES WHICH ARE OBTAINABLE ONLY ON MEDICAL PRESCRIPTION (superseding Resolution AP (82) 2) (Adopted by the Committee of Ministers on 22 September 1988 at the 419th meeting of the Ministers' Deputies) The Representatives on the Committee of Ministers of Belgium, France, the Federal Republic of Germany, Italy, Luxembourg, the Netherlands and the United Kingdom of Great Britain and Northern Ireland, these states being parties to the Partial Agreement in the social and public health field, and the Representatives of Austria, Denmark, Ireland, Spain and Switzerland, states which have participated in the public health activities carried out within the above-mentioned Partial Agreement since 1 October 1974, 2 April 1968, 23 September 1969, 21 April 1988 and 5 May 1964, respectively, Considering that the aim of the Council of Europe is to achieve greater unity between its members and that this