WO 2008/137547 Al
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Fig. L COMPOSITIONS and METHODS to INHIBIT STEM CELL and PROGENITOR CELL BINDING to LYMPHOID TISSUE and for REGENERATING GERMINAL CENTERS in LYMPHATIC TISSUES
(12) INTERNATIONAL APPLICATION PUBLISHED UNDER THE PATENT COOPERATION TREATY (PCT) (19) World Intellectual Property Organization International Bureau (10) International Publication Number (43) International Publication Date Χ 23 February 2012 (23.02.2012) WO 2U12/U24519ft ft A2 (51) International Patent Classification: AO, AT, AU, AZ, BA, BB, BG, BH, BR, BW, BY, BZ, A61K 31/00 (2006.01) CA, CH, CL, CN, CO, CR, CU, CZ, DE, DK, DM, DO, DZ, EC, EE, EG, ES, FI, GB, GD, GE, GH, GM, GT, (21) International Application Number: HN, HR, HU, ID, IL, IN, IS, JP, KE, KG, KM, KN, KP, PCT/US201 1/048297 KR, KZ, LA, LC, LK, LR, LS, LT, LU, LY, MA, MD, (22) International Filing Date: ME, MG, MK, MN, MW, MX, MY, MZ, NA, NG, NI, 18 August 201 1 (18.08.201 1) NO, NZ, OM, PE, PG, PH, PL, PT, QA, RO, RS, RU, SC, SD, SE, SG, SK, SL, SM, ST, SV, SY, TH, TJ, TM, (25) Filing Language: English TN, TR, TT, TZ, UA, UG, US, UZ, VC, VN, ZA, ZM, (26) Publication Language: English ZW. (30) Priority Data: (84) Designated States (unless otherwise indicated, for every 61/374,943 18 August 2010 (18.08.2010) US kind of regional protection available): ARIPO (BW, GH, 61/441,485 10 February 201 1 (10.02.201 1) US GM, KE, LR, LS, MW, MZ, NA, SD, SL, SZ, TZ, UG, 61/449,372 4 March 201 1 (04.03.201 1) US ZM, ZW), Eurasian (AM, AZ, BY, KG, KZ, MD, RU, TJ, TM), European (AL, AT, BE, BG, CH, CY, CZ, DE, DK, (72) Inventor; and EE, ES, FI, FR, GB, GR, HR, HU, IE, IS, ΓΓ, LT, LU, (71) Applicant : DEISHER, Theresa [US/US]; 1420 Fifth LV, MC, MK, MT, NL, NO, PL, PT, RO, RS, SE, SI, SK, Avenue, Seattle, WA 98101 (US). -

DRUG and MEDICAL DEVICE HIGHLIGHTS Helping You Maintain and Improve Your Health 2019
DRUG AND MEDICAL DEVICE HIGHLIGHTS Helping you maintain and improve your health 2019 DRUG AND MEDICAL DEVICE HIGHLIGHTS 2019 Helping you maintain and improve your health Learn about the new drugs and medical devices that Health Canada approved for sale in Canada, the information we published about potential safety issues, and our other accomplishments in 2019. Health Canada is the federal department responsible for helping the people of Canada maintain and improve their health. Health Canada is committed to improving the lives of all of Canada’s people and to making this country’s population among the healthiest in the world as measured by longevity, lifestyle and effective use of the public health care system. Également disponible en français sous le titre : Préserver et améliorer votre santé : Faits saillants sur les médicaments et les instruments médicaux 2019 To obtain additional information, please contact: Health Canada Address Locator 0900C2 Ottawa, ON K1A 0K9 Tel.: 613-957-2991 Toll free: 1-866-225-0709 Fax: 613-941-5366 TTY: 1-800-465-7735 E-mail: [email protected] © Her Majesty the Queen in Right of Canada, as represented by the Minister of Health, 2020 Publication date: May 2020 This publication may be reproduced for personal or internal use only without permission provided the source is fully acknowledged. Cat.: H161-11E-PDF ISSN: 2562-9816 Pub.: 190484 CONTENTS WELCOME TO OUR 2019 HIGHLIGHTS REPORT ..........................................................................................1 MESSAGE FROM THE CHIEF MEDICAL -

Two Cases of Immediate Stent Fracture After Zotarolimus-Eluting
Case Report http://dx.doi.org/10.4070/kcj.2015.45.1.67 Print ISSN 1738-5520 • On-line ISSN 1738-5555 Korean Circulation Journal Two Cases of Immediate Stent Fracture after Zotarolimus-Eluting Stent Implantation Pil Hyung Lee, MD, Seung-Whan Lee, MD, Jong-Young Lee, MD, Young-Hak Kim, MD, Cheol Whan Lee, MD, Duk-Woo Park, MD, Seong-Wook Park, MD, and Seung-Jung Park, MD Department of Cardiology, University of Ulsan College of Medicine, Asan Medical Center, Seoul, Korea Drug-eluting stent (DES) implantation is currently the standard treatment for various types of coronary artery disease. However, previous reports indicate that stent fractures, which usually occur after a period of time from the initial DES implantation, have increased during the DES era; stent fractures can contribute to unfavorable events such as in-stent restenosis and stent thrombosis. In our present report, we describe two cases of zotarolimus-eluting stent fracture: one that was detected six hours after implementation, and the other case that was detected immediately after deployment. Both anatomical and technical risk factors contributed to these unusual cases of imme- diate stent fracture. (Korean Circ J 2015;45(1):67-70) KEY WORDS: Drug-eluting stents; Percutaneous coronary intervention; Complications. Introduction eluting stent (ZES). Stent fractures have recently become an important concern in Cases the medical community due to their potential association with se- rious conditions such as in-stent restenosis (ISR) and stent throm- Case 1 bosis after drug-eluting stent (DES) implantation.1) Most currently A 62-year-old man, who had undergone cardiac transplantation reported stent fractures are found in lesions implanted with siroli- for advanced heart failure, was referred to our catheterization labo- mus-eluting stents (SES), likely due to the inherent platform material ratory for a regular surveillance coronary angiogram and concomi- and design of such stents.2) Furthermore, while the exact timing of tant intravascular ultrasound (IVUS) imaging. -

Related Kinases (VRK) Bound to Small-Molecule Inhibitors Identifies Different P-Loop Conformations
Structural characterization of human Vaccinia- Related Kinases (VRK) bound to small-molecule inhibitors identifies different P-loop conformations Rafael M. Couñago1,2*, Charles K. Allerston3, Pavel Savitsky3, Hatylas Azevedo4, Paulo H Godoi1,5, Carrow I. Wells6, Alessandra Mascarello4, Fernando H. de Souza Gama4, Katlin B. Massirer1,2, William J. Zuercher6, Cristiano R.W. Guimarães4 and Opher Gileadi1,3 1. Structural Genomics Consortium, Universidade Estadual de Campinas — UNICAMP, Campinas, SP, Brazil. 2. Centro de Biologia Molecular e Engenharia Genética, Universidade Estadual de Campinas, Campinas, SP, Brazil. 3. Structural Genomics Consortium and Target Discovery Institute, Nuffield Department of Clinical Medicine, University of Oxford, UK. 4. Aché Laboratórios Farmacêuticos SA, Guarulhos, SP, Brazil. 5. Department of Biochemistry and Tissue Biology, Institute of Biology, State University of Campinas, Campinas, Brazil. 6. Structural Genomics Consortium, UNC Eshelman School of Pharmacy, University of North Carolina at Chapel Hill, NC, USA. *Correspondence to Rafael M. Couñago: [email protected] (RMC) Supplementary information 1 SUPPLEMENTARY METHODS PKIS results analyses - hierarchical cluster analysis (HCL) A hierarchical clustering (HCL) analysis was performed to group kinases based on their inhibition patterns across the compounds. The average distance clustering method was employed, using sample tree selection and sample leaf order optimization. The distance metric used was the Pearson correlation and the HCL analysis was performed in the TmeV software 1. SUPPLEMENTARY REFERENCES 1 Saeed, A. I. et al. TM4: a free, open-source system for microarray data management and analysis. BioTechniques 34, 374-378, (2003). SUPPLEMENTARY FIGURES LEGENDS Supplementary Figure S1: Hierarchical clustering analysis of PKIS data. Hierarchical clustering analysis of PKIS data. -
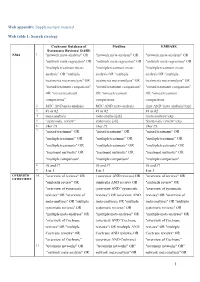
1 Web Appendix: Supplementary Material Web Table 1
Web appendix: Supplementary material Web table 1: Search strategy Cochrane Database of Medline EMBASE Systematic Reviews/ DARE NMA 1 "network meta-analysis" OR "network meta-analysis" OR "network meta-analysis" OR "network meta-regression" OR "network meta-regression" OR "network meta-regression" OR "multiple treatment meta- "multiple treatment meta- "multiple treatment meta- analysis” OR "multiple analysis OR "multiple analysis OR "multiple treatments meta-analysis" OR treatments meta-analysis" OR treatments meta-analysis" OR "mixed treatment comparison" "mixed treatment comparison" "mixed treatment comparison" OR "mixed treatment OR "mixed treatment OR "mixed treatment comparisons” comparisons comparisons 2 MTC AND meta-analysis MTC AND meta-analysis (mtc AND 'meta analysis'/exp) 3 #1 or #2 #1 or #2 #1 or #2 4 meta-analysis meta-analysis[sb] 'meta analysis'/exp 5 “systematic review” systematic [sb] 'Systematic review '/exp 6 #4or #5 #4or #5 #4or #5 7 "mixed treatment" OR "mixed treatment" OR "mixed treatment" OR "multiple treatment" OR "multiple treatment" OR "multiple treatment" OR "multiple treatments" OR "multiple treatments" OR "multiple treatments" OR "treatment networks" OR "treatment networks" OR "treatment networks" OR "multiple comparison" "multiple comparison" "multiple comparison" 8 #6 and #7 #6 and #7 #6 and #7 9 8 or 3 8 or 3 8 or 3 OVERVIEW 10 "overview of reviews" OR ( overview AND reviews) OR "overview of reviews" OR OF REVIEWS "umbrella review" OR (umbrella AND review) OR "umbrella review" OR "overview of systematic -

That Are Not Lielu Uutuullittu
THAT ARE NOT LIELUUS009987356B2 UUTUULLITTU (12 ) United States Patent ( 10 ) Patent No. : US 9 ,987 , 356 B2 Reimann et al. ( 45) Date of Patent : Jun . 5 , 2018 (54 ) ANTI -CD40 ANTIBODIES AND METHODS 762 A 12 / 1997 Queen et al. 5 , 801, 227 A 9 / 1998 Fanslow , III et al. OF ADMINISTERING THEREOF 5 , 874 ,082 A 2 / 1999 de Boer 6 ,004 , 552 A 12 / 1999 de Boer et al. ( 75 ) Inventors: Keith A . Reimann , Marblehead , MA 6 ,051 ,228 A 4 / 2000 Aruffo et al. (US ) ; Rijian Wang , Saugus, MA (US ) ; 6 ,054 ,297 A 4 / 2000 Carter et al . Christian P . Larsen , Atlanta , GA (US ) 6 ,056 , 959 A 5 /2000 de Boer et al. 6 , 132 , 978 A 10 /2000 Gelfand et al. 6 , 280 , 957 B18 / 2001 Sayegh et al. ( 73) Assignees : Beth Israel Deaconess Medical 6 , 312 ,693 B1 11/ 2001 Aruffo et al. Center , Inc ., Boston , MA (US ) ; Emory 6 , 315 , 998 B111 / 2001 de Boer et al. University , Atlanta , GA (US ) 6 , 413 ,514 B1 7 / 2002 Aruffo et al. 6 , 632, 927 B2 10 /2003 Adair et al. ( * ) Notice : Subject to any disclaimer , the term of this 7 ,063 , 845 B2 6 / 2006 Mikayama et al. patent is extended or adjusted under 35 7 , 193 , 064 B2 3 / 2007 Mikayama et al. 7 , 288 , 251 B2 10 / 2007 Bedian et al . U . S . C . 154 ( b ) by 464 days . 7 , 361 , 345 B2 4 /2008 de Boer et al. 7 , 445 , 780 B2 11/ 2008 Chu et al. (21 ) Appl . No. -

WHO Drug Information Vol. 20, No. 3, 2006
WHO DRUG INFORMATION VOLUME 20• NUMBER 3 • 2006 RECOMMENDED INN LIST 56 INTERNATIONAL NONPROPRIETARY NAMES FOR PHARMACEUTICAL SUBSTANCES WORLD HEALTH ORGANIZATION • GENEVA WHO Drug Information Vol 20, No. 3, 2006 World Health Organization WHO Drug Information Contents Biomedicines and Vaccines Counterfeit taskforce launched 192 Fake artesunate warning sheet 193 Monoclonal antibodies: a special Resolution on counterfeiting 193 regulatory challenge? 165 Improving world health through regulation of biologicals 173 Regulatory Action and News Influenza virus vaccines for 2006–2007 Safety and Efficacy Issues northern hemisphere 196 Trastuzumab approved for primary Global progress in monitoring immuniza- breast cancer 196 tion adverse events 180 Emergency contraception over-the- Intracranial haemorrhage in patients counter 197 receiving tipranavir 180 Ocular Fusarium infections: ReNu Infliximab: hepatosplenic T cell MoistureLoc® voluntary withdrawal 197 lymphoma 181 Saquinavir: withdrawal of soft gel Lamotrigine: increased risk of non- capsule Fortovase® 197 syndromic oral clefts 181 Latest list of prequalified products and Biphosphonates: osteonecrosis of manufacturers 198 the jaw 181 Clopidogrel: new medical use 199 Enoxaparin dosage in chronic kidney Dronedarone: withdrawal of marketing disease 182 authorization 199 Terbinafine and life-threatening blood dyscrasias 183 Adverse reactions in children: why Recent Publications, report? 183 Hepatitis B reactivation and anti-TNF- Information and Events alpha agents 185 MSF issues ninth antiretroviral -

Adverse Events: the Role of Formulations and Delivery Systems
12 Adverse events: the role of formulations and delivery systems 12.1 Introduction 482 12.11 Reactions to impurities 494 12.2 Excipient effects 483 12.12 Crystallisation 503 12.3 E-numbers 485 12.13 Abnormal bioavailability and adverse events 504 12.4 Cross-reactivity of drugs 487 12.14 Photochemical reactions and 12.5 Non-ionic surfactants 487 photoinduced reactions 506 12.6 Polyoxyethylene glycols 488 12.15 Conclusions 509 12.7 Adjuvants as therapeutic substances 488 References 510 12.8 Active excipients in multiple therapies 490 Further reading 511 12.9 Influence of dosage form type 491 12.10 Tear films and eye drops 492 The main purpose of formulations is to deliver active substances in accurate doses in medicines that are stable and of a high and consistent quality. Drugs are often the smallest proportion of a medicine, and the variety of other ingredients within most dose forms is large. Some of these excipients serve more than one purpose and some have some biological activity. The reason we discuss in this textbook adverse events which may follow from administration of medicines is that some of the problems that arise are the result of excipients interacting physically or chemically with drugs or with themselves, or creating instability or, in some instances, having biological activity of their own. That activity may not be a direct pharmacological or toxicological action, though some agents, such as some non-ionic surfactants, may cause anaphylactic responses or affect drug absorption rates and extent by solubilisation effects or by acting on P-glycoprotein receptors, thus influencing bioavailability. -

Percutaneous Coronary Intervention: Safety of Methotrexate and Its Possible Benefits on Restenosis After Bare-Metal Stent Deployment
Elmer ress Original Article Cardiol Res. 2016;7(3):104-109 Percutaneous Coronary Intervention: Safety of Methotrexate and Its Possible Benefits on Restenosis After Bare-Metal Stent Deployment Viviane Gouveiaa, Dinaldo C. Oliveiraa, b, Emmanuele Tenorioa, Norma Britoa, Emanuel Sarinhoa Abstract Introduction Background: Percutaneous coronary intervention (PCI) revolu- Percutaneous coronary intervention (PCI) revolutionized treat- tionized treatment of coronary artery disease. Drug-eluting stents ment of coronary artery disease (CAD); to date, it is the most are effective and safe but their cost is high, especially for some commonly used myocardial revascularization method in car- countries. The primary objective was to evaluate the safety of diology [1-3]. methotrexate (MTX) in patients who underwent PCI and the sec- Drug-eluting stents (DESs) can markedly reduce resteno- ondary goal was to evaluate the possibility that MTX has an impact sis and have become the most commonly used devices in inter- on restenosis. ventional cardiology for treatment of coronary stenosis [1]. Al- though this technique does not completely abolish restenosis, Methods: This was a transversal, prospective and descriptive study a drastic reduction due to significant decrease in neointimal that recruited 16 patients in whom PCI was planned. MTX was ad- hyperplasia has been reported [4]. ministered to patients at a dose of 5 mg/week for 2 weeks before On the other hand, the rate of restenosis bare-metal stent PCI and 8 weeks after PCI. Bare-metal stent (BMS) deployment was (BMS) varies according to clinical setting, patient and angio- performed according to standard practice. Patients were monitored graphic characteristics, with rates as high as 60% [5, 6]. -

Practical Guidance for the Evaluation and Management of Drug Hypersensitivity: Specific Drugs
Specific Drugs Practical Guidance for the Evaluation and Management of Drug Hypersensitivity: Specific Drugs Chief Editors: Ana Dioun Broyles, MD, Aleena Banerji, MD, and Mariana Castells, MD, PhD Ana Dioun Broyles, MDa, Aleena Banerji, MDb, Sara Barmettler, MDc, Catherine M. Biggs, MDd, Kimberly Blumenthal, MDe, Patrick J. Brennan, MD, PhDf, Rebecca G. Breslow, MDg, Knut Brockow, MDh, Kathleen M. Buchheit, MDi, Katherine N. Cahill, MDj, Josefina Cernadas, MD, iPhDk, Anca Mirela Chiriac, MDl, Elena Crestani, MD, MSm, Pascal Demoly, MD, PhDn, Pascale Dewachter, MD, PhDo, Meredith Dilley, MDp, Jocelyn R. Farmer, MD, PhDq, Dinah Foer, MDr, Ari J. Fried, MDs, Sarah L. Garon, MDt, Matthew P. Giannetti, MDu, David L. Hepner, MD, MPHv, David I. Hong, MDw, Joyce T. Hsu, MDx, Parul H. Kothari, MDy, Timothy Kyin, MDz, Timothy Lax, MDaa, Min Jung Lee, MDbb, Kathleen Lee-Sarwar, MD, MScc, Anne Liu, MDdd, Stephanie Logsdon, MDee, Margee Louisias, MD, MPHff, Andrew MacGinnitie, MD, PhDgg, Michelle Maciag, MDhh, Samantha Minnicozzi, MDii, Allison E. Norton, MDjj, Iris M. Otani, MDkk, Miguel Park, MDll, Sarita Patil, MDmm, Elizabeth J. Phillips, MDnn, Matthieu Picard, MDoo, Craig D. Platt, MD, PhDpp, Rima Rachid, MDqq, Tito Rodriguez, MDrr, Antonino Romano, MDss, Cosby A. Stone, Jr., MD, MPHtt, Maria Jose Torres, MD, PhDuu, Miriam Verdú,MDvv, Alberta L. Wang, MDww, Paige Wickner, MDxx, Anna R. Wolfson, MDyy, Johnson T. Wong, MDzz, Christina Yee, MD, PhDaaa, Joseph Zhou, MD, PhDbbb, and Mariana Castells, MD, PhDccc Boston, Mass; Vancouver and Montreal, -

Drug-Eluting Biostable and Erodible Stents
Journal of Controlled Release 193 (2014) 188–201 Contents lists available at ScienceDirect Journal of Controlled Release journal homepage: www.elsevier.com/locate/jconrel Review Drug-eluting biostable and erodible stents Yingying Huang, Herr Cheun Anthony Ng, Xu Wen Ng, Venkatraman Subbu ⁎ School of Materials Science and Engineering, Nanyang Technological University, Singapore 639798, Singapore article info abstract Article history: This paper reviews the latest research and development of drug-eluting stents. The emphasis is on coronary Received 26 February 2014 stenting, and both biostable and bioerodible stents are covered in this review. The advantages and shortcomings Accepted 7 May 2014 of the bioactive molecules used in these stents are analyzed, along with the rationale for using bioerodible Available online 17 May 2014 coatings. The overall emphasis is on the performance of these stents in the clinic. Based on the evaluation of the different stent types, we conclude that fully-erodible stents with a coating of antiproliferative drug will slowly Keywords: Biostable drug eluting stent (BDES) gain market share in the near future, and that the search for a more selective anti-proliferative compound will Erodible drug eluting stent (EDES) continue. Dual-drug eluting stents (DDESs) will have their market share but possibly a much smaller one than Dual-drug eluting stent (DDES) that for single-drug eluting stents due to the complexities and costs of DDES unless significantly superior Drug release kinetics performance is demonstrated in the clinic. © 2014 Elsevier B.V. All rights reserved. Contents 1. Introduction.............................................................. 189 2. Biostabledrugelutingstents(BDESs)................................................... 189 2.1. Mechanismofactionofdrugs................................................... 189 2.1.1. -

WO 2017/197056 Al 16 November 2017 (16.11.2017) W !P O PCT
(12) INTERNATIONAL APPLICATION PUBLISHED UNDER THE PATENT COOPERATION TREATY (PCT) (19) World Intellectual Property Organization International Bureau (10) International Publication Number (43) International Publication Date WO 2017/197056 Al 16 November 2017 (16.11.2017) W !P O PCT (51) International Patent Classification: AO, AT, AU, AZ, BA, BB, BG, BH, BN, BR, BW, BY, BZ, A61K 47/48 (2006.01) C07D 471/14 (2006.01) CA, CH, CL, CN, CO, CR, CU, CZ, DE, DJ, DK, DM, DO, C07D 471/04 (2006.01) DZ, EC, EE, EG, ES, FI, GB, GD, GE, GH, GM, GT, HN, HR, HU, ID, IL, IN, IR, IS, JP, KE, KG, KH, KN, KP, KR, (21) International Application Number: KW, KZ, LA, LC, LK, LR, LS, LU, LY, MA, MD, ME, MG, PCT/US20 17/032053 MK, MN, MW, MX, MY, MZ, NA, NG, NI, NO, NZ, OM, (22) International Filing Date: PA, PE, PG, PH, PL, PT, QA, RO, RS, RU, RW, SA, SC, 10 May 2017 (10.05.2017) SD, SE, SG, SK, SL, SM, ST, SV, SY,TH, TJ, TM, TN, TR, TT, TZ, UA, UG, US, UZ, VC, VN, ZA, ZM, ZW. (25) Filing Language: English (84) Designated States (unless otherwise indicated, for every (26) Publication Language: English kind of regional protection available): ARIPO (BW, GH, (30) Priority Data: GM, KE, LR, LS, MW, MZ, NA, RW, SD, SL, ST, SZ, TZ, 62/334,41 1 10 May 2016 (10.05.2016) US UG, ZM, ZW), Eurasian (AM, AZ, BY, KG, KZ, RU, TJ, TM), European (AL, AT, BE, BG, CH, CY, CZ, DE, DK, (71) Applicant: C4 THERAPEUTICS, INC.