What Would You Do with a Fluorescent Green Pig: How Novel
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-
U.S. V. Bayer AG and Monsanto Company Comment: the Sierra Club
ATTN: Kathleen S. O'Neill Chief, Transportation, Energy & Agriculture Section Antitrust Division United States Department of Justice 450 5th Street, NW, Suite 800 Washington, DC 20530 Petition in opposition to proposed U.S. v. Bayer AG and Monsanto Company settlement and merger: A merger of agrochemical giants Bayer and Monsanto would create the world's largest seed and pesticide maker. I am afraid this move will reduce competition, raise prices for consumers and farmers, and result in an unacceptable degree of control over the agricultural industry and our food supply. I am very concerned about pollinators and the increased risks to bees, butterflies and birds with the increase of Bayer's neonicotinoids. Both companies produce corn products engineered to imply the use of harmful pesticides they manufacture. The production of corn uses high amounts of nitrogen- based fertilizers and the excess sediment is contaminating our waterways, therefore I am deeply worried about increased corn production from this merger. The heavy nutrient runoff from corn is widely attributed to exacerbating the marine "Dead Zone" in the Gulf of Mexico, in which algal blooms create hypoxic conditions wherein oxygen concentration is in such low levels that marine life suffocates and dies. I urge the Department of Justice to do more prevent the Bayer-Monsanto seed and pesticide platform from growing too strong by stopping this merger. If this merger is allowed, it should require more pesticide and seed divestments in order to protect our agriculture and food supply. This merger is anti-competition, if it is approved it will fail to protect farmers, consumers and the environment by allowing further consolidation of the industrial agriculture sector. -

THE CASE for FIRST AMENDMENT PROTECTION of BIOART in 2000
FREEDOM TO EXPRESS A BIOLOGICAL CONSTRUCT: THE CASE FOR FIRST AMENDMENT PROTECTION OF BIOART KEITH SYVERSON INTRODUCTION In 2000, contemporary artist Eduardo Kac made international headlines by partnering with scientists at the Institut National de la Recherche Agronomique (INRA), France where together they genetically engineered an albino rabbit to express an enhanced green fluorescent protein (GFP)1 so that the rabbit would glow green when exposed to blue light.2 The entire project from its conception to the final display was intended as an artistic experiment to challenge society’s views on animal experimentation and the selective breeding of pets.3 This type of work that merges art and biology is called “BioArt.” According to Eduardo Kac, BioArt “employs one or more of the following approaches: (1) the coaching of biomaterials into specific inert shapes or behaviors; (2) the unusual or subversive use of biotech tools and processes; (3) the invention or transformation of living organisms with or without social environmental integration."4 BioArt has arguably existed for centuries.5 For example, some commentators have described the primitive methods of brewing beer in the Old Kingdom as form of BioArt.6 Similarly, the artificial selection involved in agriculture has been viewed by some as a form of 1 Frances Stracey, Bio-art: The Ethics Behind the Aesthetics, 10 NATURE REVIEWS: MOLECULAR CELL BIOLOGY 496, 499 (2009). 2 Id. 3 Id. 4 Eduardo Kac, Art That Looks You in the Eye: Hybrids, Clones, Mutants, Synthetics, and Transgenics, in SIGNS OF LIFE 1, 18 (Eduardo Kac ed., MIT Press, 2007). 5 See Edgar DaSilva, Art, Biotechnology and the Culture of Peace, 7 ELECTRONIC JOURNAL OF BIOTECHNOLOGY 130, 131-32 (2004) (arguing that the brewing reliefs in Egypt during the Old Kingdom from 2650 BC are an example of BioArt). -

The Era of Corporate Consolidation and the End of Competition Bayer-Monsanto, Dow-Dupont, and Chemchina-Syngenta
Research Brief October 2018 The Era of Corporate Consolidation and the End of Competition Bayer-Monsanto, Dow-DuPont, and ChemChina-Syngenta DISRUPT ECOSYSTEM ACCLERATE MONOPOLY THE EFFECTS OF CORPORATE CONSOLIDATION UNDERMINE FOOD SECURITY HARM SMALL PRODUCERS HAASINSTITUTE.BERKELEY.EDU This publication is published by the Haas Institute for a Fair and Inclusive Society at UC Berkeley This research brief is part of the Haas Institute's Shahidi Project from the Global Justice Program. The Shahidi Project (Shahidi is a Swahili word meaning “witness”) intends to demystify the power structures and capacities of transnational food and agricultural corporations within our food system. To that end, researchers have developed a robust database focusing on ten of the largest food and agricultural corporations in the world. See more at haasinstitute.berkeley.edu/shahidi. About the Authors Copyeditor Support Elsadig Elsheikh is the director Marc Abizeid Special thanks to the Food of the Global Justice program and Farm Communications at the Haas Institute for a Infographics Fund, which provided the seed Fair and Inclusive Society at Samir Gambhir funding for the Shahidi project. the University of California- Berkeley, where he oversees Report Citation Contact the program’s projects and Elsadig Elsheikh and Hossein 460 Stephens Hall research on corporate power, Ayazi. “The Era of Corporate Berkeley, CA 94720-2330 food system, forced migration, Consolidation and The End of Tel 510-642-3326 human rights, Islamophobia, Competition: Bayer-Monsanto, haasinstitute.berkeley.edu structural marginality and Dow-DuPont, and ChemChina- inclusion, and trade and Syngenta.” Haas Institute for development. a Fair and Inclusive Society at the University of California, Hossein Ayazi, PhD, is a Berkeley, CA. -
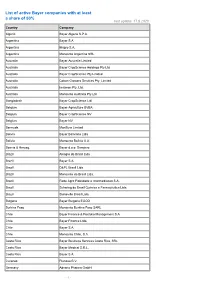
List of Active Bayer Companies with at Least a Share of 50% Last Update: 17.8.2020 Country Company
List of active Bayer companies with at least a share of 50% last update: 17.8.2020 Country Company Algeria Bayer Algerie S.P.A. Argentina Bayer S.A. Argentina Biagro S.A. Argentina Monsanto Argentina SRL Australia Bayer Australia Limited Australia Bayer CropScience Holdings Pty Ltd Australia Bayer CropScience Pty Limited Australia Cotton Growers Services Pty. Limited Australia Imaxeon Pty. Ltd. Australia Monsanto Australia Pty Ltd Bangladesh Bayer CropScience Ltd. Belgium Bayer Agriculture BVBA Belgium Bayer CropScience NV Belgium Bayer NV Bermuda MonSure Limited Bolivia Bayer Boliviana Ltda Bolivia Monsanto Bolivia S.A. Bosnia & Herzeg. Bayer d.o.o. Sarajevo Brazil Alkagro do Brasil Ltda Brazil Bayer S.A. Brazil D&PL Brasil Ltda Brazil Monsanto do Brasil Ltda. Brazil Rede Agro Fidelidade e Intermediacao S.A. Brazil Schering do Brasil Química e Farmacêutica Ltda. Brazil Stoneville Brasil Ltda. Bulgaria Bayer Bulgaria EOOD Burkina Faso Monsanto Burkina Faso SARL Chile Bayer Finance & Portfolio Management S.A. Chile Bayer Finance Ltda. Chile Bayer S.A. Chile Monsanto Chile, S.A. Costa Rica Bayer Business Services Costa Rica, SRL Costa Rica Bayer Medical S.R.L. Costa Rica Bayer S.A. Curacao Pianosa B.V. Germany Adverio Pharma GmbH - 1 - List of active Bayer companies with at least a share of 50% last update: 17.8.2020 Country Company Germany AgrEvo Verwaltungsgesellschaft mbH Germany Alcafleu Management GmbH & Co. KG Germany BGI Deutschland GmbH Germany Bayer 04 Immobilien GmbH Germany Bayer 04 Leverkusen Fußball GmbH Germany Bayer 04 Leverkusen -

Whither Plant Genetic Engineering? Allow Crops to Tolerate Environmental Stress Such As Drought, Cold, Salt, Heat, Or flood
PLANT TREK TO BOLDLY GO WHERE NO PLANT HAS GONE BEFORE On the Past, Present & Future of Plant Genetic Engineering by Richard G. Stout A HowPlantsWork.com eBook Copyright © 2013 by Richard G. Stout Version 1.0.1 PDF August, 2013 Table of Contents Preface Chapter 1: Where Do New Plants Come From? Chapter 2: How To Make A Transgenic Plant Chapter 3: Gene Guns, Terminators & Traitors Chapter 4: Farmaceuticals, Plantibodies & Edible Vaccines Chapter 5: Into The Wild Chapter 6: Are GM Plants Self-Replicating Inventions? Chapter 7: Plant Trek - The Next Generation Chapter 8: DIY Plant Genetic Engineering? Attributions About The Author Glossary about where plant biotechnology may be headed in the future, Preface including how plant biotechnology “hobbyists” may be getting into the act. Who is this book for? Please Note: This book is NOT a comprehensive textbook on plant genetic engineering and biotechnology. (If you’re looking This book is intended for people who may be curious about for such books, I’m sure you can find them at your local college plant genetic engineering, but who don’t want to read a long, bookstore or at an online bookseller.) Nor is this book meant to technical textbook on the subject. (There are provided, be a defense of genetically-engineered organisms (GMOs), however, ample links to books and articles - and also to online though I’m sure some readers will think so. Maybe here’s why. resources - for further reading.) If you’re looking for small “tastes” of information regarding various aspects of plant Since I was a graduate student in the 1970s at the University of genetic engineering, then this little book maybe just the Washington where some of the original work on transgenic informational “snack” that you’re looking for. -

Genetically Modified, Red Fluorescent Zebrafish: Detection, Crossing
Journal of Environmental Biotechnology Vol. 8, No. 2, 105–110, 2008 Original paper (regular paper) Genetically Modifi ed, Red Fluorescent Zebrafi sh: Detection, Crossing, Inheritance of Red Fluorescence, and Tolerance to Low Temperatures KIMIKO AMANUMA*, NOBUYOSHI NAKAJIMA, AKIKO H. HASHIMOTO and YASUNOBU AOKI National Institute for Environmental Studies, 16–2 Onogawa, Tsukuba, Ibaraki 305–8506, Japan * TEL: +81–(0)29–850–2390 FAX: +81–(0)29–850–2588 * E-mail: [email protected] (Received; 28 September, 2008/Accepted; 10 October, 2008) Under Cartagena Protocol domestic law, genetically modifi ed (GM) fi sh have not yet received permission to be imported or sold in Japan. However, it is anticipated that ornamental, fl uorescent GM fi sh will be imported and sold prior to any offi cial process for evaluating their eff ects on biological diversity as GM organisms (GMO). Therefore, we have devised a method for detecting GM fi sh that utilizes PCR for the detection of replication origins of plasmids that generally exist as transgenes integrated in the GMOs’ chromosomes. Using red fl uorescent zebrafi sh (rfZF), sold in Japan, and rpsL GM ZF established by us, we demonstrated the feasibility of this method for detecting GMO. Next, we examined the biological characteristics of the GM rfZF. They were able to cross with non-GM ZF; the red fl uorescence was inherited by their progeny according to expected outcomes, based on Mendelian genetics. Examination of the low temperature tolerance of rfZF and non-GM fi sh indicated that the lethal, minimum temperature was the same for both fi sh (5°C). -

Genome As a Tool of Genetic Engineering: Application in Plant and Plant Derived Medicine
International Journal of Biotech Trends and Technology (IJBTT) – Volume 8 Issue 1- Jan - March 2018 Genome as a Tool of Genetic Engineering: Application in Plant and Plant Derived Medicine A.B.M. Sharif Hossain1,2 Musamma M. Uddin2 1Department of Biology, Faculty of Science, Hail University, Hail, KSA 2Biotechnology Program, Institute of Biological Sciences, Faculty of Science, University of Malaya, 50603, Kuala Lumpur, Malaysia introduce point mutations. Genetically modified Abstract organism (GMO) is considered as an organism that The study was conducted from different is generated through genetic engineering. The first modern research data to review the innovative GMOs were bacteria in 1973, GM mice were generated in 1974 [4]. Insulin-producing bacteria latest technology in the genomics and its were commercialized in 1982 and genetically application in Agriculture, biomedicinae and modified food has been sold since 1994. Glofish, the plant derived medicine. Application of genome first GMO designed as a pet, was first sold in the in genetic engineering and molecular United States December in 2003 [4]. Genetic biotechnology have been exhibited well. engineering biotechnology has been applied in Genetically Modified Organism (GMO), numerous fields including agriculture, industrial Agrobacterium mediated recombination (T- biotechnology, and medicine. Enzymes used in DNA) and genetic engineering using molecular laundry detergent and medicines such as insulin and Biotechnology in plant, medicine and human growth hormone are now manufactured in biomedicine have been highlighted from GM cells, experimental GM cell lines and GM animals such as mice or zebra fish are being used for technology based different research data. research purposes, and genetically modified Moreover, molecular biotechnology in crops have been commercialized [4]. -

Agribusiness and Antitrust: the Bayer-Monsanto Merger, Its Legality, and Its Effect on the United States and European Union
The Global Business Law Review Volume 7 Issue 1 Article 9 7-1-2018 Agribusiness and Antitrust: The Bayer-Monsanto Merger, Its Legality, and Its Effect on the United States and European Union Aleah Douglas Cleveland-Marshall College of Law Follow this and additional works at: https://engagedscholarship.csuohio.edu/gblr Part of the Antitrust and Trade Regulation Commons, and the Business Organizations Law Commons How does access to this work benefit ou?y Let us know! Recommended Citation Aleah Douglas, Agribusiness and Antitrust: The Bayer-Monsanto Merger, Its Legality, and Its Effect on the United States and European Union, 7 Global Bus. L. Rev. 156 (2018) available at https://engagedscholarship.csuohio.edu/gblr/vol7/iss1/9 This Note is brought to you for free and open access by the Journals at EngagedScholarship@CSU. It has been accepted for inclusion in The Global Business Law Review by an authorized editor of EngagedScholarship@CSU. For more information, please contact [email protected]. AGRIBUSINESS AND ANTITRUST: THE BAYER-MONSANTO MERGER, ITS LEGALITY, AND ITS EFFECT ON THE UNITED STATES AND EUROPEAN UNION ALEAH DOUGLAS I. INTRODUCTION……………………………………………………………………... 157 II. BACKGROUND…………………………………………………………………….....158 A. A Review of the Bayer-Monsanto Merger………………………….…………... 158 B. United States Antitrust Laws…………………………………………………… 161 1. The Applicable Laws……………………………………………………. 161 2. Problems and Antitrust Violations……………………………………… 166 3. American Agribusiness…………………………………………………. 167 C. European Union Antitrust Laws……………………………………………….. 172 1. The European Union……………………………………………………. 172 2. The Applicable Laws………………………………….………………… 172 3. Problems and Antitrust Violations……………………….………....…... 174 4. European Union Agribusiness…………….…………………………….. 176 D. Illegality and Detriment …………………………………….…………………. 178 III. CONCLUSION……………………………………………………………………....... 180 ABSTRACT This note examines the current and historical antitrust laws of the United States and the European Union as they relate to the currently pending merger between Bayer and Monsanto. -

The Bio Revolution: Innovations Transforming and Our Societies, Economies, Lives
The Bio Revolution: Innovations transforming economies, societies, and our lives economies, societies, our and transforming Innovations Revolution: Bio The The Bio Revolution Innovations transforming economies, societies, and our lives May 2020 McKinsey Global Institute Since its founding in 1990, the McKinsey Global Institute (MGI) has sought to develop a deeper understanding of the evolving global economy. As the business and economics research arm of McKinsey & Company, MGI aims to help leaders in the commercial, public, and social sectors understand trends and forces shaping the global economy. MGI research combines the disciplines of economics and management, employing the analytical tools of economics with the insights of business leaders. Our “micro-to-macro” methodology examines microeconomic industry trends to better understand the broad macroeconomic forces affecting business strategy and public policy. MGI’s in-depth reports have covered more than 20 countries and 30 industries. Current research focuses on six themes: productivity and growth, natural resources, labor markets, the evolution of global financial markets, the economic impact of technology and innovation, and urbanization. Recent reports have assessed the digital economy, the impact of AI and automation on employment, physical climate risk, income inequal ity, the productivity puzzle, the economic benefits of tackling gender inequality, a new era of global competition, Chinese innovation, and digital and financial globalization. MGI is led by three McKinsey & Company senior partners: co-chairs James Manyika and Sven Smit, and director Jonathan Woetzel. Michael Chui, Susan Lund, Anu Madgavkar, Jan Mischke, Sree Ramaswamy, Jaana Remes, Jeongmin Seong, and Tilman Tacke are MGI partners, and Mekala Krishnan is an MGI senior fellow. -

Guide to Biotechnology 2008
guide to biotechnology 2008 research & development health bioethics innovate industrial & environmental food & agriculture biodefense Biotechnology Industry Organization 1201 Maryland Avenue, SW imagine Suite 900 Washington, DC 20024 intellectual property 202.962.9200 (phone) 202.488.6301 (fax) bio.org inform bio.org The Guide to Biotechnology is compiled by the Biotechnology Industry Organization (BIO) Editors Roxanna Guilford-Blake Debbie Strickland Contributors BIO Staff table of Contents Biotechnology: A Collection of Technologies 1 Regenerative Medicine ................................................. 36 What Is Biotechnology? .................................................. 1 Vaccines ....................................................................... 37 Cells and Biological Molecules ........................................ 1 Plant-Made Pharmaceuticals ........................................ 37 Therapeutic Development Overview .............................. 38 Biotechnology Industry Facts 2 Market Capitalization, 1994–2006 .................................. 3 Agricultural Production Applications 41 U.S. Biotech Industry Statistics: 1995–2006 ................... 3 Crop Biotechnology ...................................................... 41 U.S. Public Companies by Region, 2006 ........................ 4 Forest Biotechnology .................................................... 44 Total Financing, 1998–2007 (in billions of U.S. dollars) .... 4 Animal Biotechnology ................................................... 45 Biotech -

Chapter 2: Benefits and Risks of Gene Technology in Agriculture
2 %HQHILWVDQGULVNVRIJHQHWHFKQRORJ\LQ DJULFXOWXUH Introduction 2.1 Using biotechnology can be seen as extending earlier methods of plant and animal breeding which date back many thousands of years (Table 2.1).1 The technology obtains results more rapidly, is more precise, and gives access to a broader genetic base than traditional breeding techniques. These are the features that recommend its use so powerfully to plant and animal breeders. It provides an important tool when integrated with traditional breeding approaches. 2.2 The precision that gene technology offers is possible because the exact segment of a chromosome that determines a desired trait can be identified. With this capacity, traditional breeding programs can be fast tracked by locating seeds or offspring at an early stage, through gene marker technology, and breeding only from them. The Cattle Council of Australia commented on the dramatic increases in precision of genetic improvement that is possible as a result.2 In addition, genes can be removed from one organism and inserted into another. 2.3 Transgenesis, in which genes are moved from one species or organism to another, allows beneficial genes from any source to be transferred to other species or organisms. The Cooperative Research Centre (CRC) for Tropical Plant Pathology pointed out that, while conventional breeding programs have improved the pest and disease resistance of Australian crops, there 1 C Hudson, 'How industry adopts new technology', Gene Technology and Food, National Science & Industry Forum Report, Australian Academy of Science, April 1999, p. 12; Nugrain, Submission no. 25, p. 6. 2 Cattle Council of Australia, Submission no. -

Go with the Glow: Fluorescent Proteins to Light Transgenic Organisms
Review TRENDS in Biotechnology Vol.24 No.4 April 2006 Go with the glow: fluorescent proteins to light transgenic organisms C. Neal Stewart, Jr University of Tennessee, Department of Plant Sciences, Knoxville, TN 37996 USA Once a biological novelty known for their role in zebrafish [8] and the frog Xenopus laevis [9] were bioluminescence, fluorescent proteins (FPs) from marine transformed with GFP variants yielding visible green invertebrates have revolutionized the life sciences. fluorescence. FPs quickly transcended science to be used Organisms from all kingdoms have been transformed in ornamental fish and even iconic works of art. The most with the Aequorea victoria green fluorescent protein infamous of these is ‘Alba’, the GFP bunny commissioned (GFP), and biotechnology has been advanced by the use by the artist Eduardo Kac. The erstwhile albino rabbit was of FPs. This article reviews the current uses of FPs in engineered with GFP to yield a striking fluorescent whole transgenic organisms and genomics and looks phenotype (Figure 1a), which was a focal point of the beyond GFP to the complete color palette and spectral Eighth Day art exhibit, where art and biotechnology properties afforded by FPs from other marine organisms. intersected (www.ekac.org/gfpbunny.html#gfpbunnyan- Coupled with electronic devices for visualizing and chor). The transgenic rhesus monkey ‘ANDi’ [10] was quantifying FPs, recently cloned FP genes might be also iconic (at least from a news perspective) not because it useful for the ecological monitoring of transgenic had a macroscopically fluorescent body but simply because organisms in the environment. Therefore, this review it was the first reported GFP transgenic primate – also addresses the in vivo labeling of organisms with an obviously reporters and the public had a closer taxonomic emphasis on plants.