Insulin-Like Growth Factors: Actions on the Skeleton
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Osteocyte Dysfunction Promotes Osteoarthritis Through MMP13-Dependent Suppression of Subchondral Bone Homeostasis
Bone Research www.nature.com/boneres ARTICLE OPEN Osteocyte dysfunction promotes osteoarthritis through MMP13-dependent suppression of subchondral bone homeostasis Courtney M. Mazur1,2, Jonathon J. Woo 1, Cristal S. Yee1, Aaron J. Fields 1, Claire Acevedo1,3, Karsyn N. Bailey1,2, Serra Kaya1, Tristan W. Fowler1, Jeffrey C. Lotz1,2, Alexis Dang1,4, Alfred C. Kuo1,4, Thomas P. Vail1 and Tamara Alliston1,2 Osteoarthritis (OA), long considered a primary disorder of articular cartilage, is commonly associated with subchondral bone sclerosis. However, the cellular mechanisms responsible for changes to subchondral bone in OA, and the extent to which these changes are drivers of or a secondary reaction to cartilage degeneration, remain unclear. In knee joints from human patients with end-stage OA, we found evidence of profound defects in osteocyte function. Suppression of osteocyte perilacunar/canalicular remodeling (PLR) was most severe in the medial compartment of OA subchondral bone, with lower protease expression, diminished canalicular networks, and disorganized and hypermineralized extracellular matrix. As a step toward evaluating the causality of PLR suppression in OA, we ablated the PLR enzyme MMP13 in osteocytes while leaving chondrocytic MMP13 intact, using Cre recombinase driven by the 9.6-kb DMP1 promoter. Not only did osteocytic MMP13 deficiency suppress PLR in cortical and subchondral bone, but it also compromised cartilage. Even in the absence of injury, osteocytic MMP13 deficiency was sufficient to reduce cartilage proteoglycan content, change chondrocyte production of collagen II, aggrecan, and MMP13, and increase the 1234567890();,: incidence of cartilage lesions, consistent with early OA. Thus, in humans and mice, defects in PLR coincide with cartilage defects. -

Initial Sequencing and Comparative Analysis of the Mouse Genome
articles Initial sequencing and comparative analysis of the mouse genome Mouse Genome Sequencing Consortium* *A list of authors and their af®liations appears at the end of the paper ........................................................................................................................................................................................................................... The sequence of the mouse genome is a key informational tool for understanding the contents of the human genome and a key experimental tool for biomedical research. Here, we report the results of an international collaboration to produce a high-quality draft sequence of the mouse genome. We also present an initial comparative analysis of the mouse and human genomes, describing some of the insights that can be gleaned from the two sequences. We discuss topics including the analysis of the evolutionary forces shaping the size, structure and sequence of the genomes; the conservation of large-scale synteny across most of the genomes; the much lower extent of sequence orthology covering less than half of the genomes; the proportions of the genomes under selection; the number of protein-coding genes; the expansion of gene families related to reproduction and immunity; the evolution of proteins; and the identi®cation of intraspecies polymorphism. With the complete sequence of the human genome nearly in hand1,2, covering about 90% of the euchromatic human genome, with about the next challenge is to extract the extraordinary trove of infor- 35% in ®nished form1. Since then, progress towards a complete mation encoded within its roughly 3 billion nucleotides. This human sequence has proceeded swiftly, with approximately 98% of information includes the blueprints for all RNAs and proteins, the genome now available in draft form and about 95% in ®nished the regulatory elements that ensure proper expression of all genes, form. -

Osteocyte Differentiation and the Formation of an Interconnected Cellular Network in Vitro
EuropeanMJ Mc Garrigle Cells and et alMaterials. Vol. 31 2016 (pages 323-340) DOI: 10.22203/eCM.v031a21Formation of an interconnected osteocyte ISSN 1473-2262 network OSTEOCYTE DIFFERENTIATION AND THE FORMATION OF AN INTERCONNECTED CELLULAR NETWORK IN VITRO M.J. Mc Garrigle1, C.A. Mullen1, M.G. Haugh1, M.C. Voisin1 and L.M. McNamara1* 1 Centre for Biomechanics Research (BMEC), Biomedical Engineering, College of Engineering and Informatics, National University of Ireland, Galway Abstract Introduction Extracellular matrix (ECM) stiffness and cell density can In bone, osteocyte cells form a complex three-dimensional regulate osteoblast differentiation in two dimensional (3D) communication network that plays a vital role in environments. However, it is not yet known how maintaining bone health by monitoring physical cues osteoblast-osteocyte differentiation is regulated within a arising during load-bearing activity and directing the 3D ECM environment, akin to that existing in vivo. In this activity of osteoblasts and osteoclasts to initiate bone study we test the hypothesis that osteocyte differentiation is formation and resorption (Burger and Klein-Nulend, 1999). regulated by a 3D cell environment, ECM stiffness and cell Osteocytes are formed when cuboidal-like osteoblasts density. We encapsulated MC3T3-E1 pre-osteoblastic cells become embedded within soft secreted osteoid and start to at varied cell densities (0.25, 1 and 2 × 106 cells/mL) within change morphologically to a dendritic shape characteristic microbial transglutaminase (mtgase) gelatin hydrogels of an osteocyte. This transition is accompanied by a loss of low (0.58 kPa) and high (1.47 kPa) matrix stiffnesses. of cell volume (reduced organelle content) (Knothe Tate Cellular morphology was characterised from phalloidin- et al., 2004; Palumbo et al., 2004) and an increase in the FITC and 4’,6-diamidino-2-phenylindole (DAPI) dilactate formation and elongation of thin cytoplasmic projections staining. -

Associated 16P11.2 Deletion in Drosophila Melanogaster
ARTICLE DOI: 10.1038/s41467-018-04882-6 OPEN Pervasive genetic interactions modulate neurodevelopmental defects of the autism- associated 16p11.2 deletion in Drosophila melanogaster Janani Iyer1, Mayanglambam Dhruba Singh1, Matthew Jensen1,2, Payal Patel 1, Lucilla Pizzo1, Emily Huber1, Haley Koerselman3, Alexis T. Weiner 1, Paola Lepanto4, Komal Vadodaria1, Alexis Kubina1, Qingyu Wang 1,2, Abigail Talbert1, Sneha Yennawar1, Jose Badano 4, J. Robert Manak3,5, Melissa M. Rolls1, Arjun Krishnan6,7 & 1234567890():,; Santhosh Girirajan 1,2,8 As opposed to syndromic CNVs caused by single genes, extensive phenotypic heterogeneity in variably-expressive CNVs complicates disease gene discovery and functional evaluation. Here, we propose a complex interaction model for pathogenicity of the autism-associated 16p11.2 deletion, where CNV genes interact with each other in conserved pathways to modulate expression of the phenotype. Using multiple quantitative methods in Drosophila RNAi lines, we identify a range of neurodevelopmental phenotypes for knockdown of indi- vidual 16p11.2 homologs in different tissues. We test 565 pairwise knockdowns in the developing eye, and identify 24 interactions between pairs of 16p11.2 homologs and 46 interactions between 16p11.2 homologs and neurodevelopmental genes that suppress or enhance cell proliferation phenotypes compared to one-hit knockdowns. These interac- tions within cell proliferation pathways are also enriched in a human brain-specific network, providing translational relevance in humans. Our study indicates a role for pervasive genetic interactions within CNVs towards cellular and developmental phenotypes. 1 Department of Biochemistry and Molecular Biology, The Pennsylvania State University, University Park, PA 16802, USA. 2 Bioinformatics and Genomics Program, The Huck Institutes of the Life Sciences, The Pennsylvania State University, University Park, PA 16802, USA. -

MECHANISMS in ENDOCRINOLOGY: Novel Genetic Causes of Short Stature
J M Wit and others Genetics of short stature 174:4 R145–R173 Review MECHANISMS IN ENDOCRINOLOGY Novel genetic causes of short stature 1 1 2 2 Jan M Wit , Wilma Oostdijk , Monique Losekoot , Hermine A van Duyvenvoorde , Correspondence Claudia A L Ruivenkamp2 and Sarina G Kant2 should be addressed to J M Wit Departments of 1Paediatrics and 2Clinical Genetics, Leiden University Medical Center, PO Box 9600, 2300 RC Leiden, Email The Netherlands [email protected] Abstract The fast technological development, particularly single nucleotide polymorphism array, array-comparative genomic hybridization, and whole exome sequencing, has led to the discovery of many novel genetic causes of growth failure. In this review we discuss a selection of these, according to a diagnostic classification centred on the epiphyseal growth plate. We successively discuss disorders in hormone signalling, paracrine factors, matrix molecules, intracellular pathways, and fundamental cellular processes, followed by chromosomal aberrations including copy number variants (CNVs) and imprinting disorders associated with short stature. Many novel causes of GH deficiency (GHD) as part of combined pituitary hormone deficiency have been uncovered. The most frequent genetic causes of isolated GHD are GH1 and GHRHR defects, but several novel causes have recently been found, such as GHSR, RNPC3, and IFT172 mutations. Besides well-defined causes of GH insensitivity (GHR, STAT5B, IGFALS, IGF1 defects), disorders of NFkB signalling, STAT3 and IGF2 have recently been discovered. Heterozygous IGF1R defects are a relatively frequent cause of prenatal and postnatal growth retardation. TRHA mutations cause a syndromic form of short stature with elevated T3/T4 ratio. Disorders of signalling of various paracrine factors (FGFs, BMPs, WNTs, PTHrP/IHH, and CNP/NPR2) or genetic defects affecting cartilage extracellular matrix usually cause disproportionate short stature. -

Neurologic Outcomes in Friedreich Ataxia: Study of a Single-Site Cohort E415
Volume 6, Number 3, June 2020 Neurology.org/NG A peer-reviewed clinical and translational neurology open access journal ARTICLE Neurologic outcomes in Friedreich ataxia: Study of a single-site cohort e415 ARTICLE Prevalence of RFC1-mediated spinocerebellar ataxia in a North American ataxia cohort e440 ARTICLE Mutations in the m-AAA proteases AFG3L2 and SPG7 are causing isolated dominant optic atrophy e428 ARTICLE Cerebral autosomal dominant arteriopathy with subcortical infarcts and leukoencephalopathy revisited: Genotype-phenotype correlations of all published cases e434 Academy Officers Neurology® is a registered trademark of the American Academy of Neurology (registration valid in the United States). James C. Stevens, MD, FAAN, President Neurology® Genetics (eISSN 2376-7839) is an open access journal published Orly Avitzur, MD, MBA, FAAN, President Elect online for the American Academy of Neurology, 201 Chicago Avenue, Ann H. Tilton, MD, FAAN, Vice President Minneapolis, MN 55415, by Wolters Kluwer Health, Inc. at 14700 Citicorp Drive, Bldg. 3, Hagerstown, MD 21742. Business offices are located at Two Carlayne E. Jackson, MD, FAAN, Secretary Commerce Square, 2001 Market Street, Philadelphia, PA 19103. Production offices are located at 351 West Camden Street, Baltimore, MD 21201-2436. Janis M. Miyasaki, MD, MEd, FRCPC, FAAN, Treasurer © 2020 American Academy of Neurology. Ralph L. Sacco, MD, MS, FAAN, Past President Neurology® Genetics is an official journal of the American Academy of Neurology. Journal website: Neurology.org/ng, AAN website: AAN.com CEO, American Academy of Neurology Copyright and Permission Information: Please go to the journal website (www.neurology.org/ng) and click the Permissions tab for the relevant Mary E. -

Genome-Wide Transcriptional Sequencing Identifies Novel Mutations in Metabolic Genes in Human Hepatocellular Carcinoma DAOUD M
CANCER GENOMICS & PROTEOMICS 11 : 1-12 (2014) Genome-wide Transcriptional Sequencing Identifies Novel Mutations in Metabolic Genes in Human Hepatocellular Carcinoma DAOUD M. MEERZAMAN 1,2 , CHUNHUA YAN 1, QING-RONG CHEN 1, MICHAEL N. EDMONSON 1, CARL F. SCHAEFER 1, ROBERT J. CLIFFORD 2, BARBARA K. DUNN 3, LI DONG 2, RICHARD P. FINNEY 1, CONSTANCE M. CULTRARO 2, YING HU1, ZHIHUI YANG 2, CU V. NGUYEN 1, JENNY M. KELLEY 2, SHUANG CAI 2, HONGEN ZHANG 2, JINGHUI ZHANG 1,4 , REBECCA WILSON 2, LAUREN MESSMER 2, YOUNG-HWA CHUNG 5, JEONG A. KIM 5, NEUNG HWA PARK 6, MYUNG-SOO LYU 6, IL HAN SONG 7, GEORGE KOMATSOULIS 1 and KENNETH H. BUETOW 1,2 1Center for Bioinformatics and Information Technology, National Cancer Institute, Rockville, MD, U.S.A.; 2Laboratory of Population Genetics, National Cancer Institute, National Cancer Institute, Bethesda, MD, U.S.A.; 3Basic Prevention Science Research Group, Division of Cancer Prevention, National Cancer Institute, Bethesda, MD, U.S.A; 4Department of Biotechnology/Computational Biology, St. Jude Children’s Research Hospital, Memphis, TN, U.S.A.; 5Department of Internal Medicine, University of Ulsan College of Medicine, Asan Medical Center, Seoul, Korea; 6Department of Internal Medicine, University of Ulsan College of Medicine, Ulsan University Hospital, Ulsan, Korea; 7Department of Internal Medicine, College of Medicine, Dankook University, Cheon-An, Korea Abstract . We report on next-generation transcriptome Worldwide, liver cancer is the fifth most common cancer and sequencing results of three human hepatocellular carcinoma the third most common cause of cancer-related mortality (1). tumor/tumor-adjacent pairs. -

IGFBP3 Goat Polyclonal Antibody – TA303275 | Origene
OriGene Technologies, Inc. 9620 Medical Center Drive, Ste 200 Rockville, MD 20850, US Phone: +1-888-267-4436 [email protected] EU: [email protected] CN: [email protected] Product datasheet for TA303275 IGFBP3 Goat Polyclonal Antibody Product data: Product Type: Primary Antibodies Applications: WB Recommended Dilution: ELISA: 1:64,000. WB: 0.02-0.06µg/ml. Reactivity: Human (Expected from sequence similarity: Mouse, Rat, Dog) Host: Goat Isotype: IgG Clonality: Polyclonal Immunogen: Peptide with sequence C-RYKVDYESQSTDTQN, from the internal region of the protein sequence according to NP_001013416.1; NP_000589.2. Formulation: Supplied at 0.5 mg/ml in Tris saline, 0.02% sodium azide, pH7.3 with 0.5% bovine serum albumin. Purification: Purified from goat serum by ammonium sulphate precipitation followed by antigen affinity chromatography using the immunizing peptide. Supplied at 0.5 mg/ml in Tris saline, 0.02% sodium azide, pH7.3 with 0.5% bovine serum albumin. Aliquot and store at -20°C. Minimize freezing and thawing. Conjugation: Unconjugated Storage: Store at -20°C as received. Stability: Stable for 12 months from date of receipt. Gene Name: insulin like growth factor binding protein 3 Database Link: NP_000589 Entrez Gene 16009 MouseEntrez Gene 24484 RatEntrez Gene 100855619 DogEntrez Gene 3486 Human P17936 This product is to be used for laboratory only. Not for diagnostic or therapeutic use. View online » ©2021 OriGene Technologies, Inc., 9620 Medical Center Drive, Ste 200, Rockville, MD 20850, US 1 / 2 IGFBP3 Goat Polyclonal Antibody – TA303275 Background: This gene is a member of the insulin-like growth factor binding protein (IGFBP) family and encodes a protein with an IGFBP domain and a thyroglobulin type-I domain. -
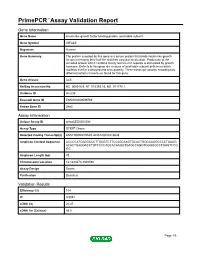
Primepcr™Assay Validation Report
PrimePCR™Assay Validation Report Gene Information Gene Name insulin-like growth factor binding protein, acid labile subunit Gene Symbol IGFALS Organism Human Gene Summary The protein encoded by this gene is a serum protein that binds insulin-like growth factors increasing their half-life and their vascular localization. Production of the encoded protein which contains twenty leucine-rich repeats is stimulated by growth hormone. Defects in this gene are a cause of acid-labile subunit deficiency which maifests itself in a delayed and slow puberty. Three transcript variants encoding two different isoforms have been found for this gene. Gene Aliases ALS RefSeq Accession No. NC_000016.9, NT_010393.16, NG_011778.1 UniGene ID Hs.839 Ensembl Gene ID ENSG00000099769 Entrez Gene ID 3483 Assay Information Unique Assay ID qHsaCED0003592 Assay Type SYBR® Green Detected Coding Transcript(s) ENST00000215539, ENST00000415638 Amplicon Context Sequence ACCCCATCAGGCCCTTGCGTCTTCCAGCAAGTGCACTGGGCAGGCCCCTGAGG ACACTGAGGACCTGTCCCCAGCACAAGGTGAGCCAGGTGGGGGCCTGAGTCCG GG Amplicon Length (bp) 78 Chromosome Location 16:1840473-1840580 Assay Design Exonic Purification Desalted Validation Results Efficiency (%) 104 R2 0.9991 cDNA Cq 26.47 cDNA Tm (Celsius) 85.5 Page 1/5 PrimePCR™Assay Validation Report gDNA Cq 24.22 Specificity (%) 100 Information to assist with data interpretation is provided at the end of this report. Page 2/5 PrimePCR™Assay Validation Report IGFALS, Human Amplification Plot Amplification of cDNA generated from 25 ng of universal reference RNA Melt -

Cartilage & Bone
Cartilage & bone Red: important. Black: in male|female slides. Gray: notes|extra. Editing File ➢ OBJECTIVES • describe the microscopic structure, distribution and growth of the different types of Cartilage • describe the microscopic structure, distribution and growth of the different types of Bone Histology team 437 | MSK block | Lecture one ➢ REMEMBER from last block (connective tissue lecture) Components of connective tissue Fibers Cells Collagenous, Matrix difference elastic & the intercellular substance, in which types reticular cells and fibers are embedded. Rigid (rubbery, Hard (solid) Fluid (liquid) Soft firm) “Cartilage” “Bone” “Blood” “C.T Proper” Histology team 437 | MSK block | Lecture one CARTILAGE “Chondro- = relating to cartilage” BONE “Osteo- = relating to bone” o Its specialized type of connective tissue with a rigid o Its specialized type of connective tissue with a hard matrix (ﻻ يكسر بسهولة) matrix o Its usually nonvascular (avascular = lack of blood o Types: vessels) 1) Compact bone 2) Spongy bone o Its poor nerve supply o Components: 1) Bone cells: o All cartilage contain collagen fiber type II • Osteogenic cells • Osteoblasts o Types: • Osteocytes 1) Hyaline cartilage (main type) • Osteoclasts 2) Elastic cartilage 2) Bone Matrix (calcified osteoid tissue): 3) Fibrocartilage • hard because it is calcified (Calcium salts) • It contains collagen fibers type I • It forms bone lamellae and trabeculae 3) Periosteum 4) Endosteum o Functions: 1) body support 2) protection of vital organs as brain & bone marrow 3) calcium -

The Osteocyte: from “Prisoner” to “Orchestrator”
Journal of Functional Morphology and Kinesiology Review The Osteocyte: From “Prisoner” to “Orchestrator” Carla Palumbo * and Marzia Ferretti Section of Human Morphology, Department of Biomedical, Metabolic and Neural Sciences, University of Modena and Reggio Emilia, 41124 Modena, Italy; [email protected] * Correspondence: [email protected] Abstract: Osteocytes are the most abundant bone cells, entrapped inside the mineralized bone matrix. They derive from osteoblasts through a complex series of morpho-functional modifications; such modifications not only concern the cell shape (from prismatic to dendritic) and location (along the vascular bone surfaces or enclosed inside the lacuno-canalicular cavities, respectively) but also their role in bone processes (secretion/mineralization of preosseous matrix and/or regulation of bone remodeling). Osteocytes are connected with each other by means of different types of junctions, among which the gap junctions enable osteocytes inside the matrix to act in a neuronal-like manner, as a functional syncytium together with the cells placed on the vascular bone surfaces (osteoblasts or bone lining cells), the stromal cells and the endothelial cells, i.e., the bone basic cellular system (BBCS). Within the BBCS, osteocytes can communicate in two ways: by means of volume transmission and wiring transmission, depending on the type of signals (metabolic or mechanical, respectively) received and/or to be forwarded. The capability of osteocytes in maintaining skeletal and mineral homeostasis is due to the fact that it acts as a mechano-sensor, able to transduce mechanical strains into biological signals and to trigger/modulate the bone remodeling, also because of the relevant role of sclerostin secreted by osteocytes, thus regulating different bone cell signaling pathways. -

Constitutional Delay of Growth and Puberty Is Not Commonly Associated
European Journal of Endocrinology (2008) 158 473–477 ISSN 0804-4643 CLINICAL STUDY Constitutional delay of growth and puberty is not commonly associated with mutations in the acid labile subunit gene I Banerjee1, D Hanson2, R Perveen2, A Whatmore3, G C Black2 and P E Clayton1,3 1Department of Paediatric Endocrinology, Royal Manchester Children’s Hospital, Pendlebury, Manchester M27 4HA, UK, 2Academic Unit of Medical Genetics and 3Endocrine Science Research Group, University of Manchester, Manchester M13 9NT, UK (Correspondence should be addressed to I Banerjee; Email: [email protected]) Abstract Objectives: Constitutional delay of growth and puberty (CDGP) is a common clinical condition that may be inherited as an autosomal dominant, recessive or X-linked trait. However, single-gene defects underlying CDGP have not yet been identified. A small number of children (to date 10) with modest growth failure and in the majority delayed puberty, a phenotype similar to that of CDGP, have been reported to carry mutations in the IGF acid labile subunit (IGFALS) gene which encodes the ALS, a part of the ternary complex carrying IGF-I in the circulation. The aim of our study was to screen a well- characterised CDGP cohort exhibiting a range of growth retardation and pubertal delay for pathogenic sequence variants in IGFALS. Design and methods: We used denaturing high performance liquid chromatography (dHPLC) to screen for IGFALS mutations in DNA samples from 90 children (80 males) with CDGP of predominantly White European origin. DNA fragments generating abnormal waveforms were directly sequenced. Results:NoIGFALS mutation was identified in the coding sequences or exon–intron boundaries in our CDGP cohort.