Science Journals
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Research Article Label-Free Proteomics of the Fetal Pancreas Identifies Deficits in the Peroxisome in Rats with Intrauterine Growth Restriction
Hindawi Oxidative Medicine and Cellular Longevity Volume 2019, Article ID 1520753, 15 pages https://doi.org/10.1155/2019/1520753 Research Article Label-Free Proteomics of the Fetal Pancreas Identifies Deficits in the Peroxisome in Rats with Intrauterine Growth Restriction Xiaomei Liu ,1 Yanyan Guo ,1 Jun Wang ,1,2 Linlin Gao ,3 and Caixia Liu 1 1Key Laboratory of Maternal-Fetal Medicine of Liaoning Province, Department of Obstetrics and Gynecology, Shengjing Hospital of China Medical University, Shenyang 110004, China 2Department of Obstetrics and Gynecology, Benxi Central Hospital of China Medical University, Benxi 117022, China 3Medical Research Center, Shengjing Hospital, China Medical University, Shenyang 110004, China Correspondence should be addressed to Xiaomei Liu; [email protected] Received 14 May 2019; Revised 31 August 2019; Accepted 9 September 2019; Published 3 November 2019 Guest Editor: Roberta Cascella Copyright © 2019 Xiaomei Liu et al. This is an open access article distributed under the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited. Aim. The objective of the present study was to identify differentially expressed proteins (DEPs) in the pancreas of a fetus with intrauterine growth restriction (IUGR) and to investigate the molecular mechanisms leading to adulthood diabetes in IUGR. Methods. The IUGR rat model was induced by maternal protein malnutrition. The fetal pancreas was collected at embryonic day 20 (E20). Protein was extracted, pooled, and subjected to label-free quantitative proteomic analysis. Bioinformatics analysis (GO and IPA) was performed to define the pathways and networks associated with DEPs. LC-MS results were confirmed by western blotting and/or quantitative PCR (q-PCR). -

Peroxisomal Disorders and Their Mouse Models Point to Essential Roles of Peroxisomes for Retinal Integrity
International Journal of Molecular Sciences Review Peroxisomal Disorders and Their Mouse Models Point to Essential Roles of Peroxisomes for Retinal Integrity Yannick Das, Daniëlle Swinkels and Myriam Baes * Lab of Cell Metabolism, Department of Pharmaceutical and Pharmacological Sciences, KU Leuven, 3000 Leuven, Belgium; [email protected] (Y.D.); [email protected] (D.S.) * Correspondence: [email protected] Abstract: Peroxisomes are multifunctional organelles, well known for their role in cellular lipid homeostasis. Their importance is highlighted by the life-threatening diseases caused by peroxisomal dysfunction. Importantly, most patients suffering from peroxisomal biogenesis disorders, even those with a milder disease course, present with a number of ocular symptoms, including retinopathy. Patients with a selective defect in either peroxisomal α- or β-oxidation or ether lipid synthesis also suffer from vision problems. In this review, we thoroughly discuss the ophthalmological pathology in peroxisomal disorder patients and, where possible, the corresponding animal models, with a special emphasis on the retina. In addition, we attempt to link the observed retinal phenotype to the underlying biochemical alterations. It appears that the retinal pathology is highly variable and the lack of histopathological descriptions in patients hampers the translation of the findings in the mouse models. Furthermore, it becomes clear that there are still large gaps in the current knowledge on the contribution of the different metabolic disturbances to the retinopathy, but branched chain fatty acid accumulation and impaired retinal PUFA homeostasis are likely important factors. Citation: Das, Y.; Swinkels, D.; Baes, Keywords: peroxisome; Zellweger; metabolism; fatty acid; retina M. Peroxisomal Disorders and Their Mouse Models Point to Essential Roles of Peroxisomes for Retinal Integrity. -

Supplementary Table S4. FGA Co-Expressed Gene List in LUAD
Supplementary Table S4. FGA co-expressed gene list in LUAD tumors Symbol R Locus Description FGG 0.919 4q28 fibrinogen gamma chain FGL1 0.635 8p22 fibrinogen-like 1 SLC7A2 0.536 8p22 solute carrier family 7 (cationic amino acid transporter, y+ system), member 2 DUSP4 0.521 8p12-p11 dual specificity phosphatase 4 HAL 0.51 12q22-q24.1histidine ammonia-lyase PDE4D 0.499 5q12 phosphodiesterase 4D, cAMP-specific FURIN 0.497 15q26.1 furin (paired basic amino acid cleaving enzyme) CPS1 0.49 2q35 carbamoyl-phosphate synthase 1, mitochondrial TESC 0.478 12q24.22 tescalcin INHA 0.465 2q35 inhibin, alpha S100P 0.461 4p16 S100 calcium binding protein P VPS37A 0.447 8p22 vacuolar protein sorting 37 homolog A (S. cerevisiae) SLC16A14 0.447 2q36.3 solute carrier family 16, member 14 PPARGC1A 0.443 4p15.1 peroxisome proliferator-activated receptor gamma, coactivator 1 alpha SIK1 0.435 21q22.3 salt-inducible kinase 1 IRS2 0.434 13q34 insulin receptor substrate 2 RND1 0.433 12q12 Rho family GTPase 1 HGD 0.433 3q13.33 homogentisate 1,2-dioxygenase PTP4A1 0.432 6q12 protein tyrosine phosphatase type IVA, member 1 C8orf4 0.428 8p11.2 chromosome 8 open reading frame 4 DDC 0.427 7p12.2 dopa decarboxylase (aromatic L-amino acid decarboxylase) TACC2 0.427 10q26 transforming, acidic coiled-coil containing protein 2 MUC13 0.422 3q21.2 mucin 13, cell surface associated C5 0.412 9q33-q34 complement component 5 NR4A2 0.412 2q22-q23 nuclear receptor subfamily 4, group A, member 2 EYS 0.411 6q12 eyes shut homolog (Drosophila) GPX2 0.406 14q24.1 glutathione peroxidase -

E3 Ubiquitin Ligase SP1 Regulates Peroxisome Biogenesis in Arabidopsis
E3 ubiquitin ligase SP1 regulates peroxisome PNAS PLUS biogenesis in Arabidopsis Ronghui Pana, John Satkovicha, and Jianping Hua,b,1 aDepartment of Energy Plant Research Laboratory, Michigan State University, East Lansing, MI 48824; and bPlant Biology Department, Michigan State University, East Lansing, MI 48824 Edited by Natasha V. Raikhel, Center for Plant Cell Biology, Riverside, CA, and approved September 30, 2016 (received for review August 17, 2016) Peroxisomes are ubiquitous eukaryotic organelles that play pivotal signal type 1) and N-terminal PTS2 sequences, respectively roles in a suite of metabolic processes and often act coordinately (15, 16). In Arabidopsis, PEX5 is also required for PTS2 protein with other organelles, such as chloroplasts and mitochondria. Peroxi- import (16). Two membrane proteins, PEX13 and PEX14, form somes import proteins to the peroxisome matrix by peroxins (PEX the docking site for PEX5 and PEX7 (17, 18). After receptor proteins), but how the function of the PEX proteins is regulated is docking, cargo proteins translocate into the matrix before re- poorly understood. In this study, we identified the Arabidopsis RING ceptors are recycled to the cytosol (19–21). These processes re- (really interesting new gene) type E3 ubiquitin ligase SP1 [suppressor quire the RING (really interesting new gene)-finger peroxins of plastid protein import locus 1 (ppi1) 1] as a peroxisome membrane PEX2,PEX10,andPEX12(22–25), the ATPases PEX1 and protein with a regulatory role in peroxisome protein import. SP1 PEX6 and their membrane tether APEM9 (aberrant peroxisome interacts physically with the two components of the peroxisome morphology 9) and the ubiquitin-conjugating enzyme PEX4 and protein docking complex PEX13–PEX14 and the (RING)-finger per- its membrane anchor PEX22 (26–29). -

Amplification of Glyceronephosphate O-Acyltransferase and Recruitment of USP30 Stabilize DRP1 to Promote Hepatocarcinogenesis
Author Manuscript Published OnlineFirst on August 24, 2018; DOI: 10.1158/0008-5472.CAN-18-0340 Author manuscripts have been peer reviewed and accepted for publication but have not yet been edited. Amplification of glyceronephosphate O-acyltransferase and recruitment of USP30 stabilize DRP1 to promote hepatocarcinogenesis Li Gu #, 1, 2, Yahui Zhu #, 1, 2, Xi Lin 1, 2, Yajun Li 1, 2, Kasa Cui 1, 2, Edward V. Prochownik 3, Youjun Li 1, 2,* 1 Hubei Key Laboratory of Cell Homeostasis, College of Life Sciences, Wuhan University, Wuhan 430072, China 2 Medical Research Institute, School of Medicine, Wuhan University, Wuhan 430071, China 3Division of Hematology/Oncology, Children's Hospital of Pittsburgh of UPMC, The Department of Microbiology and Molecular Genetics and The Hillman Cancer Center of UPMC The University of Pittsburgh Medical Center, Pittsburgh, Pennsylvania 15224, USA #These authors contributed equally. Running title: GNPAT and USP30-mediated DRP1 stabilization in HCC. *Correspondence to: Youjun Li, Hubei Key Laboratory of Cell Homeostasis, College of Life Sciences, Wuhan University, Wuhan 430072, China; Medical Research Institute, School of Medicine, Wuhan University, Wuhan 430071, China. Tel.: (86-27) 6875-2050; Fax: (86-27) 6875-2560; E-mail: [email protected] Conflict of interest: The authors declare no conflicts of interest. Downloaded from cancerres.aacrjournals.org on September 25, 2021. © 2018 American Association for Cancer Research. Author Manuscript Published OnlineFirst on August 24, 2018; DOI: 10.1158/0008-5472.CAN-18-0340 -

Perkinelmer Genomics to Request the Saliva Swab Collection Kit for Patients That Cannot Provide a Blood Sample As Whole Blood Is the Preferred Sample
Zellweger Spectrum Disorder Panel Test Code D4611 Test Summary This panel analyzes 15 genes that have been associated with Zellweger Spectrum Disorders. Turn-Around-Time (TAT)* 3 - 5 weeks Acceptable Sample Types Whole Blood (EDTA) (Preferred sample type) DNA, Isolated Dried Blood Spots Saliva Acceptable Billing Types Self (patient) Payment Institutional Billing Commercial Insurance Indications for Testing This panel may be appropriate for individuals with signs and symptoms of a peroxisomal biogenesis disfuction and/or Zellweger syndrome spectrum disorder and/or a family history of these conditions. Test Description This panel analyzes 15 genes that have been associated with Zellweger Spectrum Disorders. Both sequencing and deletion/duplication (CNV) analysis will be performed on the coding regions of all genes included (unless otherwise marked). All analysis is performed utilizing Next Generation Sequencing (NGS) technology. CNV analysis is designed to detect the majority of deletions and duplications of three exons or greater in size. Smaller CNV events may also be detected and reported, but additional follow-up testing is recommended if a smaller CNV is suspected. All variants are classified according to ACMG guidelines. Condition Description Zellweger syndrome spectrum disorders include Zellweger syndrome (ZS), neonatal adrenoleukodystrophy (NALD), an intermediate form and infantile Refsum disease (IRD). Symptoms may include hypotonia, feeding problems, hearing and vision loss, seizures, distinctive facial characteristics and skeletal abnormalities. Zellweger spectrum disorder is estimated to occur in 1/50,000 individuals. Genes ACOX1, AMACR, HSD17B4, PEX1, PEX10, PEX12, PEX13, PEX14, PEX16, PEX19, PEX2, PEX26, PEX3, PEX5, PEX6 Test Methods and Limitations Sequencing is performed on genomic DNA using an Agilent targeted sequence capture method to enrich for the genes on this panel. -

Mechanisms and Functions of Pexophagy in Mammalian Cells
cells Review Mechanisms and Functions of Pexophagy in Mammalian Cells Jing Li 1 and Wei Wang 2,* 1 Department of Integrated Traditional Chinese and Western Medicine, Tongji Hospital, Tongji Medical College, Huazhong University of Science and Technology, Wuhan 430030, China; [email protected] 2 Department of Human Anatomy, School of Basic Medicine, Tongji Medical College, Huazhong University of Science and Technology, Wuhan 430030, China * Correspondence: [email protected] Abstract: Peroxisomes play essential roles in diverse cellular metabolism functions, and their dy- namic homeostasis is maintained through the coordination of peroxisome biogenesis and turnover. Pexophagy, selective autophagic degradation of peroxisomes, is a major mechanism for removing damaged and/or superfluous peroxisomes. Dysregulation of pexophagy impairs the physiological functions of peroxisomes and contributes to the progression of many human diseases. However, the mechanisms and functions of pexophagy in mammalian cells remain largely unknown com- pared to those in yeast. This review focuses on mammalian pexophagy and aims to advance the understanding of the roles of pexophagy in human health and diseases. Increasing evidence shows that ubiquitination can serve as a signal for pexophagy, and ubiquitin-binding receptors, substrates, and E3 ligases/deubiquitinases involved in pexophagy have been described. Alternatively, pex- ophagy can be achieved in a ubiquitin-independent manner. We discuss the mechanisms of these ubiquitin-dependent and ubiquitin-independent pexophagy pathways and summarize several in- ducible conditions currently used to study pexophagy. We highlight several roles of pexophagy in human health and how its dysregulation may contribute to diseases. Citation: Li, J.; Wang, W. Keywords: peroxisome; autophagy; mammalian; pexophagy; ubiquitin; receptor Mechanisms and Functions of Pexophagy in Mammalian Cells. -

E3 Ubiquitin Ligase SP1 Regulates Peroxisome Biogenesis In
E3 ubiquitin ligase SP1 regulates peroxisome PNAS PLUS biogenesis in Arabidopsis Ronghui Pana, John Satkovicha, and Jianping Hua,b,1 aDepartment of Energy Plant Research Laboratory, Michigan State University, East Lansing, MI 48824; and bPlant Biology Department, Michigan State University, East Lansing, MI 48824 Edited by Natasha V. Raikhel, Center for Plant Cell Biology, Riverside, CA, and approved September 30, 2016 (received for review August 17, 2016) Peroxisomes are ubiquitous eukaryotic organelles that play pivotal signal type 1) and N-terminal PTS2 sequences, respectively roles in a suite of metabolic processes and often act coordinately (15, 16). In Arabidopsis, PEX5 is also required for PTS2 protein with other organelles, such as chloroplasts and mitochondria. Peroxi- import (16). Two membrane proteins, PEX13 and PEX14, form somes import proteins to the peroxisome matrix by peroxins (PEX the docking site for PEX5 and PEX7 (17, 18). After receptor proteins), but how the function of the PEX proteins is regulated is docking, cargo proteins translocate into the matrix before re- poorly understood. In this study, we identified the Arabidopsis RING ceptors are recycled to the cytosol (19–21). These processes re- (really interesting new gene) type E3 ubiquitin ligase SP1 [suppressor quire the RING (really interesting new gene)-finger peroxins of plastid protein import locus 1 (ppi1) 1] as a peroxisome membrane PEX2,PEX10,andPEX12(22–25), the ATPases PEX1 and protein with a regulatory role in peroxisome protein import. SP1 PEX6 and their membrane tether APEM9 (aberrant peroxisome interacts physically with the two components of the peroxisome morphology 9) and the ubiquitin-conjugating enzyme PEX4 and protein docking complex PEX13–PEX14 and the (RING)-finger per- its membrane anchor PEX22 (26–29). -

Genetic Classification and Mutational Spectrum of More Than 600 Patients
Genetic classification and mutational spectrum of more than 600 patients with a Zellweger syndrome spectrum disorder Merel Sanne Ebberink, Petra Mooyer, Jeannette Gootjes, Janet Koster, Ronald J.A. Wanders, Hans Waterham To cite this version: Merel Sanne Ebberink, Petra Mooyer, Jeannette Gootjes, Janet Koster, Ronald J.A. Wanders, et al.. Genetic classification and mutational spectrum of more than 600 patients with a Zellweger syn- drome spectrum disorder. Human Mutation, Wiley, 2010, 32 (1), pp.59. 10.1002/humu.21388. hal-00599469 HAL Id: hal-00599469 https://hal.archives-ouvertes.fr/hal-00599469 Submitted on 10 Jun 2011 HAL is a multi-disciplinary open access L’archive ouverte pluridisciplinaire HAL, est archive for the deposit and dissemination of sci- destinée au dépôt et à la diffusion de documents entific research documents, whether they are pub- scientifiques de niveau recherche, publiés ou non, lished or not. The documents may come from émanant des établissements d’enseignement et de teaching and research institutions in France or recherche français ou étrangers, des laboratoires abroad, or from public or private research centers. publics ou privés. Human Mutation Genetic classification and mutational spectrum of more than 600 patients with a Zellweger syndrome spectrum disorder For Peer Review Journal: Human Mutation Manuscript ID: humu-2010-0170.R1 Wiley - Manuscript type: Research Article Date Submitted by the 30-Aug-2010 Author: Complete List of Authors: Ebberink, Merel; Academic Medical Centre, University of Amsterdam, Laboratoy -
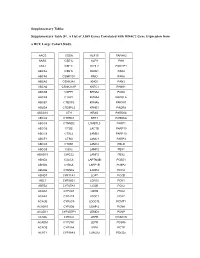
Supplementary Tables Supplementary Table S1. a List of 3,039 Genes
Supplementary Tables Supplementary Table S1. A List of 3,039 Genes Correlated with HDAC2 Gene Expression from a HCC Large Cohort Study. AACS CSDA KLF15 PAFAH2 AASS CSE1L KLF9 PAH ABAT CSE1L KLHL2 PAK1IP1 ABCA2 CSE1L KLKB1 PAK4 ABCA5 CSNK1G1 KMO PAK6 ABCA5 CSNK2A1 KNG1 PAN3 ABCA6 CSNK2A1P KNTC1 PANK1 ABCA9 CSPP1 KPNA2 PAOX ABCA9 CTBP2 KPNA4 PAPOLA ABCB1 CTDSP2 KPNA5 PAPSS1 ABCB4 CTDSPL2 KPNB1 PAQR8 ABCC10 CTH KRAS PARD6A ABCC6 CTHRC1 KRT3 PARD6G ABCC6 CTNND2 L3MBTL3 PARP1 ABCC6 CTSB LACTB PARP10 ABCC9 CTSL2 LAMB1 PARP10 ABCF1 CTSO LAMC1 PARP3 ABCG5 CTXN1 LAMC2 PBLD ABCG8 CUX2 LAMP2 PBX1 ABHD15 CWC22 LAMP2 PBX2 ABHD2 CXXC6 LAPTM4B PCBD1 ABHD5 CYB5A LARP1B PCBP2 ABHD6 CYB5D2 LARP4 PCCA ABHD7 CYP11A1 LCAT PCCB ABL1 CYP26B1 LCN12 PCK1 ABTB2 CYP27A1 LCOR PCK2 ACAA1 CYP2A7 LDHD PCK2 ACAA2 CYP2C8 LDOC1 PCK2 ACACB CYP2C9 LDOC1L PCMT1 ACAD10 CYP2D6 LEAP-2 PCNA ACAD11 CYP2D7P1 LEMD3 PCNP ACADL CYP2J2 LEPR PCSK1N ACADM CYP2W1 LEPR PCSK6 ACADS CYP3A4 LEPR PCTP ACAT1 CYP3A43 LGALS2 PDCD2 ACBD3 CYP3A5 LGALS7 PDCD2 ACBD4 CYP4A11 LIMA1 PDCL3 ACBD5 CYP4A22 LIMS2 PDE5A ACCN3 CYP4F11 LINGO1 PDK2 ACMSD CYP4F12 LIPG PDK4 ACO1 CYP4F2 LMAN1 PDS5B ACOT1 CYP4F3 LMAN2 PEA15 ACOT12 CYP7B1 LMAN2L PEBP1 ACOT4 CYP8B1 LMCD1 PECR ACOT8 DAK LMNB2 PELI1 ACOX1 DAO LOC120376 PELP1 ACOX2 DARS LOC123876 PEMT ACPL2 DBF4 LOC127295 PEPD ACSL1 DBN1 LOC130773 PER1 ACSM2A DBNDD2 LOC136143 PEX11G ACSM2B DBNL LOC143543 PEX13 ACSM2B DBT LOC143941 PEX14 ACSM2B DCAF16 LOC146439 PFDN5 ACSM3 DCAF7 LOC147710 PFKFB1 ACSM3 DCDC2 LOC158345 PGF ACSM5 DCTN2 LOC201725 PGM1 ACSM5 DCXR LOC220433 -

Cerebellar and Hepatic Alterations in ACBD5-Deficient Mice Are
ARTICLE https://doi.org/10.1038/s42003-020-01442-x OPEN Cerebellar and hepatic alterations in ACBD5- deficient mice are associated with unexpected, distinct alterations in cellular lipid homeostasis Warda Darwisch1,7, Marino von Spangenberg1,7, Jana Lehmann1, Öznur Singin1, Geralt Deubert1, Sandra Kühl1, 1234567890():,; Johannes Roos1, Heinz Horstmann2, Christoph Körber2, Simone Hoppe2, Hongwei Zheng2, Thomas Kuner2, Mia L. Pras-Raves3,4, Antoine H. C. van Kampen4,5, Hans R. Waterham3, Kathrin V. Schwarz6, Jürgen G. Okun6, ✉ Christian Schultz1, Frédéric M. Vaz 3 & Markus Islinger 1 ACBD5 deficiency is a novel peroxisome disorder with a largely uncharacterized pathology. ACBD5 was recently identified in a tethering complex mediating membrane contacts between peroxisomes and the endoplasmic reticulum (ER). An ACBD5-deficient mouse was analyzed to correlate ACBD5 tethering functions with the disease phenotype. ACBD5- deficient mice exhibit elevated very long-chain fatty acid levels and a progressive cerebellar pathology. Liver did not exhibit pathologic changes but increased peroxisome abundance and drastically reduced peroxisome-ER contacts. Lipidomics of liver and cerebellum revealed tissue-specific alterations in distinct lipid classes and subspecies. In line with the neurological pathology, unusual ultra-long chain fatty acids (C > 32) were elevated in phosphocholines from cerebelli but not liver indicating an organ-specific imbalance in fatty acid degradation and elongation pathways. By contrast, ether lipid formation was perturbed in liver towards an accumulation of alkyldiacylglycerols. The alterations in several lipid classes suggest that ACBD5, in addition to its acyl-CoA binding function, might maintain peroxisome-ER contacts in order to contribute to the regulation of anabolic and catabolic cellular lipid pathways. -

Comprehensive Proteome Analysis of Lysosomes Reveals the Diverse Function of Macrophages in Immune Responses
www.impactjournals.com/oncotarget/ Oncotarget, 2017, Vol. 8, (No. 5), pp: 7420-7440 Research Paper: Immunology Comprehensive proteome analysis of lysosomes reveals the diverse function of macrophages in immune responses Yanpan Gao1,*, Yanyu Chen1,*, Shaohua Zhan1, Wenhao Zhang2, Feng Xiong1 and Wei Ge1 1 Department of Immunology, National Key Laboratory of Medical Molecular Biology, Institute of Basic Medical Sciences, Chinese Academy of Medical Sciences, Dongcheng, Beijing, China 2 MOE Key Laboratory of Bioinformatics, School of Life Sciences, Tsinghua University, Beijing, China * These authors have contributed equally to this work Correspondence to: Feng Xiong, email: [email protected] Correspondence to: Wei Ge, email: [email protected] Keywords: innate immunity, lysosome, macrophage, quantitative proteomics, tandem mass tag labeling, Immunology and Micro- biology Section, Immune response, Immunity Received: July 14, 2016 Accepted: December 31, 2016 Published: January 08, 2017 ABSTRACT Phagocytosis and autophagy in macrophages have been shown to be essential to both innate and adaptive immunity. Lysosomes are the main catabolic subcellular organelles responsible for degradation and recycling of both extracellular and intracellular material, which are the final steps in phagocytosis and autophagy. However, the molecular mechanisms underlying lysosomal functions after infection remain obscure. In this study, we conducted a quantitative proteomics analysis of the changes in constitution and glycosylation of proteins in lysosomes derived from murine RAW 264.7 macrophage cells treated with different types of pathogens comprising examples of bacteria (Listeria monocytogenes, L. m), DNA viruses (herpes simplex virus type-1, HSV-1) and RNA viruses (vesicular stomatitis virus, VSV). In total, 3,704 lysosome-related proteins and 300 potential glycosylation sites on 193 proteins were identified.