THE IDENTIFICATION and CHARACTERIZATION of a GROUP of ER TPR-CONTAINING ADAPTER PROTEINS Johan C
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Responses of Bats to White-Nose Syndrome and Implications for Conservation
University of New Hampshire University of New Hampshire Scholars' Repository Doctoral Dissertations Student Scholarship Spring 2020 Responses of Bats to White-Nose Syndrome and Implications for Conservation Meghan Stark University of New Hampshire, Durham Follow this and additional works at: https://scholars.unh.edu/dissertation Recommended Citation Stark, Meghan, "Responses of Bats to White-Nose Syndrome and Implications for Conservation" (2020). Doctoral Dissertations. 2518. https://scholars.unh.edu/dissertation/2518 This Dissertation is brought to you for free and open access by the Student Scholarship at University of New Hampshire Scholars' Repository. It has been accepted for inclusion in Doctoral Dissertations by an authorized administrator of University of New Hampshire Scholars' Repository. For more information, please contact [email protected]. RESPONSES OF BATS TO WHITE-NOSE SYNDROME AND IMPLICATIONS FOR CONSERVATION BY MEGHAN A. STARK B.S., University of Alabama at Birmingham, 2013 DISSERTATION Submitted to the University of New Hampshire in Partial Fulfillment of the Requirements for the Degree of Doctor of Philosophy In Genetics May 2020 i This dissertation was examined and approved in partial fulfillment of the requirements for the degree of Ph.D. in Genetics by: Dissertation Director, Matthew MacManes, Assoc. Prof. UNH MCBS Jeffrey T. Foster, Associate Professor, NAU PMI W. Kelley Thomas, Professor, UNH MCBS Rebecca Rowe, Associate Professor, UNH NREN Thomas Lee, Associate Professor Emeritus, UNH NREN On April 6, 2020 Approval signatures are on file with the University of New Hampshire Graduate School. ii DEDICATION I dedicate this work to all of the strong women in my life: Myra Michele Ange Heather Michelle Coons Kaitlyn Danielle Cagle Brindlee Michelle Coons Patricia Gail Miller Sarah Jean Lane “Here’s to strong women. -

University of Copenhagen, Copenhagen, Denmark
Endoplasmic reticulum transmembrane protein TMTC3 contributes to O-mannosylation of E-cadherin, cellular adherence, and embryonic gastrulation Graham, Jill B.; Sunryd, Johan C.; Mathavan, Ketan; Weir, Emma; Larsen, Ida Signe Bohse; Halim, Adnan; Clausen, Henrik; Cousin, Hélène; Alfandari, Dominque; Hebert, Daniel N. Published in: Molecular Biology of the Cell DOI: 10.1091/mbc.E19-07-0408 Publication date: 2020 Document version Publisher's PDF, also known as Version of record Document license: CC BY-NC-SA Citation for published version (APA): Graham, J. B., Sunryd, J. C., Mathavan, K., Weir, E., Larsen, I. S. B., Halim, A., Clausen, H., Cousin, H., Alfandari, D., & Hebert, D. N. (2020). Endoplasmic reticulum transmembrane protein TMTC3 contributes to O- mannosylation of E-cadherin, cellular adherence, and embryonic gastrulation. Molecular Biology of the Cell, 31(3), 167-183. https://doi.org/10.1091/mbc.E19-07-0408 Download date: 28. Sep. 2021 ER transmembrane protein TMTC3 contributes to O-mannosylation of E-cadherin, Cellular Adherence and Embryonic Gastrulation Jill B. Graham1,3, Johan C. Sunryd1,3, Ketan Mathavan2,3, Emma Weir2,3, Ida Signe Bohse Larsen4,5, Adnan Halim4,5, Henrik Clausen4,5, Hélène Cousin2,3 Dominque Alfandari2,3 and Daniel N. Hebert1,3* 1Department of Biochemistry and Molecular Biology and 2Department of Veterinary and Animal Sciences, University of Massachusetts, Amherst, Amherst, MA USA. 3Program in Molecular and Cellular Biology, University of Massachusetts, Amherst, Amherst, MA USA. 4Department of Cellular and Molecular Medicine, University of Copenhagen, Copenhagen, Denmark. 5Copenhagen Center for Glycomics, University of Copenhagen, Copenhagen, Denmark. * To whom correspondence should be addressed: Daniel N. -

The Genetics of Bipolar Disorder
Molecular Psychiatry (2008) 13, 742–771 & 2008 Nature Publishing Group All rights reserved 1359-4184/08 $30.00 www.nature.com/mp FEATURE REVIEW The genetics of bipolar disorder: genome ‘hot regions,’ genes, new potential candidates and future directions A Serretti and L Mandelli Institute of Psychiatry, University of Bologna, Bologna, Italy Bipolar disorder (BP) is a complex disorder caused by a number of liability genes interacting with the environment. In recent years, a large number of linkage and association studies have been conducted producing an extremely large number of findings often not replicated or partially replicated. Further, results from linkage and association studies are not always easily comparable. Unfortunately, at present a comprehensive coverage of available evidence is still lacking. In the present paper, we summarized results obtained from both linkage and association studies in BP. Further, we indicated new potential interesting genes, located in genome ‘hot regions’ for BP and being expressed in the brain. We reviewed published studies on the subject till December 2007. We precisely localized regions where positive linkage has been found, by the NCBI Map viewer (http://www.ncbi.nlm.nih.gov/mapview/); further, we identified genes located in interesting areas and expressed in the brain, by the Entrez gene, Unigene databases (http://www.ncbi.nlm.nih.gov/entrez/) and Human Protein Reference Database (http://www.hprd.org); these genes could be of interest in future investigations. The review of association studies gave interesting results, as a number of genes seem to be definitively involved in BP, such as SLC6A4, TPH2, DRD4, SLC6A3, DAOA, DTNBP1, NRG1, DISC1 and BDNF. -

Discovery of an O-Mannosylation Pathway Selectively Serving Cadherins and Protocadherins
Discovery of an O-mannosylation pathway selectively serving cadherins and protocadherins Ida Signe Bohse Larsena,1, Yoshiki Narimatsua,1, Hiren Jitendra Joshia,1, Lina Siukstaitea, Oliver J. Harrisonb, Julia Braschb, Kerry M. Goodmanb, Lars Hansena, Lawrence Shapirob,c,d, Barry Honigb,c,d,e, Sergey Y. Vakhrusheva, Henrik Clausena, and Adnan Halima,2,3 aDepartment of Cellular and Molecular Medicine, Faculty of Health Sciences, Copenhagen Center for Glycomics, University of Copenhagen, DK-2200 Copenhagen, Denmark; bDepartment of Biochemistry and Molecular Biophysics, Columbia University, New York, NY 10032; cZuckerman Mind Brain Behavior Institute, Columbia University, New York, NY 10032; dDepartment of Systems Biology, Columbia University, New York, NY 10032; and eHoward Hughes Medical Institute, Columbia University, New York, NY 10032 Edited by Stuart A. Kornfeld, Washington University School of Medicine, St. Louis, MO, and approved September 6, 2017 (received for review May 22, 2017) The cadherin (cdh) superfamily of adhesion molecules carry muscular dystrophies that have been designated α-dystroglyca- O-linked mannose (O-Man) glycans at highly conserved sites nopathies because deficient O-Man glycosylation of α-DG dis- localized to specific β-strands of their extracellular cdh (EC) domains. rupts the interaction between the dystrophin glycoprotein complex These O-Man glycans do not appear to be elongated like O-Man and the ECM (7–9). Several studies have also implicated de- glycans found on α-dystroglycan (α-DG), and we recently demon- ficiency of POMT2 with E-cdh dysfunction (10–12), although di- strated that initiation of cdh/protocadherin (pcdh) O-Man glycosyl- rect evidence for a role in glycosylation of cdhs and pcdhs is ation is not dependent on the evolutionary conserved POMT1/ missing. -

TMTC4 (Y-19): Sc-84645
SAN TA C RUZ BI OTEC HNOL OG Y, INC . TMTC4 (Y-19): sc-84645 BACKGROUND PRODUCT The tetratricopeptide repeat (TPR) motif is a degenerate, 34 amino acid Each vial contains 100 µg IgG in 1.0 ml of PBS with < 0.1% sodium azide sequence found in many proteins and acts to mediate protein-protein inter- and 0.1% gelatin. actions in various pathways. At the sequence level, there can be up to Blocking peptide available for competition studies, sc-84645 P, (100 µg 16 tandem TPR repeats, each of which has a helix-turn-helix shape that stacks peptide in 0.5 ml PBS containing < 0.1% sodium azide and 0.2% BSA). on other TPR repeats to achieve ligand binding specificity. TMTC4 (trans - membrane and tetratricopeptide repeat containing 4) is a 741 amino acid APPLICATIONS multi-pass membrane protein that contains 8 TPR repeats and is expressed as multiple alternatively spliced isoforms. The gene encoding TMTC4 maps TMTC4 (Y-19) is recommended for detection of TMTC4 of mouse, rat and to human chromosome 13, which houses over 400 genes, such as BRCA2 human origin by Western Blotting (starting dilution 1:200, dilution range and RB1, and comprises nearly 4% of the human genome. 1:100-1:1000), immunoprecipitation [1-2 µg per 100-500 µg of total protein (1 ml of cell lysate)], immunofluorescence (starting dilution 1:50, dilution REFERENCES range 1:50-1:500) and solid phase ELISA (starting dilution 1:30, dilution range 1:30-1:3000). 1. Young, J.C., Obermann, W.M. and Hartl, F.U. -

Lupus Nephritis Supp Table 5
Supplementary Table 5 : Transcripts and DAVID pathways correlating with the expression of CD4 in lupus kidney biopsies Positive correlation Negative correlation Transcripts Pathways Transcripts Pathways Identifier Gene Symbol Correlation coefficient with CD4 Annotation Cluster 1 Enrichment Score: 26.47 Count P_Value Benjamini Identifier Gene Symbol Correlation coefficient with CD4 Annotation Cluster 1 Enrichment Score: 3.16 Count P_Value Benjamini ILMN_1727284 CD4 1 GOTERM_BP_FAT translational elongation 74 2.50E-42 1.00E-38 ILMN_1681389 C2H2 zinc finger protein-0.40001984 INTERPRO Ubiquitin-conjugating enzyme/RWD-like 17 2.00E-05 4.20E-02 ILMN_1772218 HLA-DPA1 0.934229063 SP_PIR_KEYWORDS ribosome 60 2.00E-41 4.60E-39 ILMN_1768954 RIBC1 -0.400186083 SMART UBCc 14 1.00E-04 3.50E-02 ILMN_1778977 TYROBP 0.933302249 KEGG_PATHWAY Ribosome 65 3.80E-35 6.60E-33 ILMN_1699190 SORCS1 -0.400223681 SP_PIR_KEYWORDS ubl conjugation pathway 81 1.30E-04 2.30E-02 ILMN_1689655 HLA-DRA 0.915891173 SP_PIR_KEYWORDS protein biosynthesis 91 4.10E-34 7.20E-32 ILMN_3249088 LOC93432 -0.400285215 GOTERM_MF_FAT small conjugating protein ligase activity 35 1.40E-04 4.40E-02 ILMN_3228688 HLA-DRB1 0.906190291 SP_PIR_KEYWORDS ribonucleoprotein 114 4.80E-34 6.70E-32 ILMN_1680436 CSH2 -0.400299744 SP_PIR_KEYWORDS ligase 54 1.50E-04 2.00E-02 ILMN_2157441 HLA-DRA 0.902996561 GOTERM_CC_FAT cytosolic ribosome 59 3.20E-33 2.30E-30 ILMN_1722755 KRTAP6-2 -0.400334007 GOTERM_MF_FAT acid-amino acid ligase activity 40 1.60E-04 4.00E-02 ILMN_2066066 HLA-DRB6 0.901531942 SP_PIR_KEYWORDS -
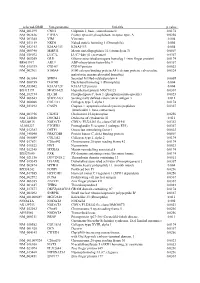
Supplementary Figures S1-S3
selected-GBID Uni-genename Uni-title p value NM_001299 CNN1 Calponin 1, basic, smooth muscle 0.0174 NM_002836 PTPRA Protein tyrosine phosphatase, receptor type, A 0.0256 NM_003380 VIM Vimentin 0.004 NM_033119 NKD1 Naked cuticle homolog 1 (Drosophila) 0.004 NM_052913 KIAA1913 KIAA1913 0.004 NM_005940 MMP11 Matrix metallopeptidase 11 (stromelysin 3) 0.0069 NM_018032 LUC7L LUC7-like (S. cerevisiae) 0.0367 NM_005269 GLI1 Glioma-associated oncogene homolog 1 (zinc finger protein) 0.0174 BE463997 ARL9 ADP-ribosylation factor-like 9 0.0367 NM_015939 CGI-09 CGI-09 protein 0.0023 NM_002961 S100A4 S100 calcium binding protein A4 (calcium protein, calvasculin, 0.0324 metastasin, murine placental homolog) NM_003014 SFRP4 Secreted frizzled-related protein 4 0.0005 NM_080759 DACH1 Dachshund homolog 1 (Drosophila) 0.004 NM_053042 KIAA1729 KIAA1729 protein 0.004 BX415194 MGC16121 Hypothetical protein MGC16121 0.0367 NM_182734 PLCB1 Phospholipase C, beta 1 (phosphoinositide-specific) 0.0023 NM_006643 SDCCAG3 Serologically defined colon cancer antigen 3 0.011 NM_000088 COL1A1 Collagen, type I, alpha 1 0.0174 NM_033292 CASP1 Caspase 1, apoptosis-related cysteine peptidase 0.0367 (interleukin 1, beta, convertase) NM_003956 CH25H Cholesterol 25-hydroxylase 0.0256 NM_144658 DOCK11 Dedicator of cytokinesis 11 0.011 AK024935 NODATA CDNA: FLJ21283 fis, clone COL01910 0.0363 AL050227 PTGER3 Prostaglandin E receptor 3 (subtype EP3) 0.0367 NM_012383 OSTF1 Osteoclast stimulating factor 1 0.0023 NM_145040 PRKCDBP Protein kinase C, delta binding protein 0.0069 NM_000089 -

Deletion of Tmtc4 Activates the Unfolded Protein Response and Causes Postnatal Hearing Loss
Deletion of Tmtc4 activates the unfolded protein response and causes postnatal hearing loss Jiang Li, … , Dylan K. Chan, Elliott H. Sherr J Clin Invest. 2018;128(11):5150-5162. https://doi.org/10.1172/JCI97498. Research Article Cell biology Otology Hearing loss is a significant public health concern, affecting over 250 million people worldwide. Both genetic and environmental etiologies are linked to hearing loss, but in many cases the underlying cellular pathophysiology is not well understood, highlighting the importance of further discovery. We found that inactivation of the gene Tmtc4 (transmembrane and tetratricopeptide repeat 4), which was broadly expressed in the mouse cochlea, caused acquired hearing loss in mice. Our data showed Tmtc4 enriched in the endoplasmic reticulum, and that it functioned by regulating Ca2+ dynamics and the unfolded protein response (UPR). Given this genetic linkage of the UPR to hearing loss, we demonstrated a direct link between the more common noise-induced hearing loss (NIHL) and the UPR. These experiments suggested a novel approach to treatment. We demonstrated that the small-molecule UPR and stress response modulator ISRIB (integrated stress response inhibitor), which activates eIF2B, prevented NIHL in a mouse model. Moreover, in an inverse genetic complementation approach, we demonstrated that mice with homozygous inactivation of both Tmtc4 and Chop had less hearing loss than knockout ofT mtc4 alone. This study implicated a novel mechanism for hearing impairment, highlighting a potential treatment approach for a broad range of human hearing loss disorders. Find the latest version: https://jci.me/97498/pdf RESEARCH ARTICLE The Journal of Clinical Investigation Deletion of Tmtc4 activates the unfolded protein response and causes postnatal hearing loss Jiang Li,1 Omar Akil,2 Stephanie L. -

Us 2018 / 0305689 A1
US 20180305689A1 ( 19 ) United States (12 ) Patent Application Publication ( 10) Pub . No. : US 2018 /0305689 A1 Sætrom et al. ( 43 ) Pub . Date: Oct. 25 , 2018 ( 54 ) SARNA COMPOSITIONS AND METHODS OF plication No . 62 /150 , 895 , filed on Apr. 22 , 2015 , USE provisional application No . 62/ 150 ,904 , filed on Apr. 22 , 2015 , provisional application No. 62 / 150 , 908 , (71 ) Applicant: MINA THERAPEUTICS LIMITED , filed on Apr. 22 , 2015 , provisional application No. LONDON (GB ) 62 / 150 , 900 , filed on Apr. 22 , 2015 . (72 ) Inventors : Pål Sætrom , Trondheim (NO ) ; Endre Publication Classification Bakken Stovner , Trondheim (NO ) (51 ) Int . CI. C12N 15 / 113 (2006 .01 ) (21 ) Appl. No. : 15 /568 , 046 (52 ) U . S . CI. (22 ) PCT Filed : Apr. 21 , 2016 CPC .. .. .. C12N 15 / 113 ( 2013 .01 ) ; C12N 2310 / 34 ( 2013. 01 ) ; C12N 2310 /14 (2013 . 01 ) ; C12N ( 86 ) PCT No .: PCT/ GB2016 /051116 2310 / 11 (2013 .01 ) $ 371 ( c ) ( 1 ) , ( 2 ) Date : Oct . 20 , 2017 (57 ) ABSTRACT The invention relates to oligonucleotides , e . g . , saRNAS Related U . S . Application Data useful in upregulating the expression of a target gene and (60 ) Provisional application No . 62 / 150 ,892 , filed on Apr. therapeutic compositions comprising such oligonucleotides . 22 , 2015 , provisional application No . 62 / 150 ,893 , Methods of using the oligonucleotides and the therapeutic filed on Apr. 22 , 2015 , provisional application No . compositions are also provided . 62 / 150 ,897 , filed on Apr. 22 , 2015 , provisional ap Specification includes a Sequence Listing . SARNA sense strand (Fessenger 3 ' SARNA antisense strand (Guide ) Mathew, Si Target antisense RNA transcript, e . g . NAT Target Coding strand Gene Transcription start site ( T55 ) TY{ { ? ? Targeted Target transcript , e . -

“Functional Characterization of the RNA Binding Protein RALY”
International Doctoral School in Biomolecular Sciences XXV Cycle “Functional characterization of the RNA binding protein RALY” Tutor Professor Paolo MACCHI CIBIO- University of Trento Ph.D. Thesis of Albertomaria MORO CIBIO- University of Trento Academic Year 2012-2013 To myself and my little world... INDEX 1 - Introduction ....................................................................................................... 1 1.1 The RNA-binding proteins ............................................................................. 2 1.2 The hnRNP super family ............................................................................... 5 1.2.1 Properties of hnRNPs ............................................................................ 5 1.3 RALY: a new member of hnRNPs ................................................................. 9 2 - Topic of my PhD project ................................................................................. 13 3 - Results ............................................................................................................ 15 3.1 Publication 1:............................................................................................... 15 3.2 additional results ......................................................................................... 16 3.2.1 RALY localization ................................................................................. 16 3.2.2 Polyribosome profiling .......................................................................... 20 3.2.3 The microarray -

Similarities in Gene Expression Profiles During in Vitro Aging of Primary Human Embryonic Lung and Foreskin Fibroblasts
Hindawi Publishing Corporation BioMed Research International Volume 2015, Article ID 731938, 17 pages http://dx.doi.org/10.1155/2015/731938 Research Article Similarities in Gene Expression Profiles during In Vitro Aging of Primary Human Embryonic Lung and Foreskin Fibroblasts Shiva Marthandan,1 Steffen Priebe,2 Mario Baumgart,1 Marco Groth,1 Alessandro Cellerino,1,3 Reinhard Guthke,2 Peter Hemmerich,1 and Stephan Diekmann1 1 Leibniz-Institute for Age Research-Fritz Lipmann Institute e.V. (FLI), 07745 Jena, Germany 2Leibniz Institute for Natural Product Research and Infection Biology-Hans-Knoll-Institute¨ e.V. (HKI), 07745 Jena, Germany 3Laboratory of Neurobiology, Scuola Normale Superiore, University of Pisa, 56126 Pisa, Italy Correspondence should be addressed to Shiva Marthandan; [email protected] Received 23 January 2015; Revised 14 June 2015; Accepted 22 June 2015 Academic Editor: Hesham H. Ali Copyright © 2015 Shiva Marthandan et al. This is an open access article distributed under the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited. Replicative senescence is of fundamental importance for the process of cellular aging, since it is a property of most of our somatic cells. Here, we elucidated this process by comparing gene expression changes, measured by RNA-seq, in fibroblasts originating from two different tissues, embryonic lung (MRC-5) and foreskin (HFF), at five different time points during their transition into senescence. Although the expression patterns of both fibroblast cell lines can be clearly distinguished, the similar differential expression of an ensemble of genes was found to correlate well with their transition into senescence, with only a minority of genes being cell line specific. -

Foxp1 in Forebrain Pyramidal Neurons Controls Gene Expression Required for Spatial Learning and Synaptic Plasticity
The Journal of Neuroscience, November 8, 2017 • 37(45):10917–10931 • 10917 Cellular/Molecular Foxp1 in Forebrain Pyramidal Neurons Controls Gene Expression Required for Spatial Learning and Synaptic Plasticity Daniel J. Araujo,1 XKazuya Toriumi,1,4 XChristine O. Escamilla,2 Ashwinikumar Kulkarni,1 Ashley G. Anderson,1 X Matthew Harper,1 XNoriyoshi Usui,1 XJacob Ellegood,5 Jason P. Lerch,5,6 XShari G. Birnbaum,3 XHaley O. Tucker,7 X Craig M. Powell,1,2,3 and XGenevieve Konopka1 1Department of Neuroscience, 2Department of Neurology, and 3Department of Psychiatry, University of Texas Southwestern Medical Center, Dallas, Texas 75390, 4Project for Schizophrenia Research, Department of Psychiatry and Behavioral Sciences, Tokyo Metropolitan Institute of Medical Science, Tokyo 156-8506, Japan, 5Mouse Imaging Centre, The Hospital for Sick Children, Toronto, Ontario M5T 3H7, Canada, 6Department of Medical Biophysics, University of Toronto, Toronto, Ontario M5G 1L7, Canada, and 7University of Texas at Austin, Section of Molecular Genetics and Microbiology, Austin, Texas 78712 Genetic perturbations of the transcription factor Forkhead Box P1 (FOXP1) are causative for severe forms of autism spectrum disorder that are often comorbid with intellectual disability. Recent work has begun to reveal an important role for FoxP1 in brain development, but the brain-region-specific contributions of Foxp1 to autism and intellectual disability phenotypes have yet to be determined fully. Here, we describe Foxp1 conditional knock-out (Foxp1 cKO) male and female mice with loss of Foxp1 in the pyramidal neurons of the neocortexandtheCA1/CA2subfieldsofthehippocampus.Foxp1 cKO miceexhibitbehavioralphenotypesthatareofpotentialrelevancetoautism spectrum disorder, including hyperactivity, increased anxiety, communication impairments, and decreased sociability. In addition, Foxp1 cKO mice have gross deficits in learning and memory tasks of relevance to intellectual disability.