Hypermethioninemia Leads to Fatal Bleeding and Increased Mortality in a Transgenic I278T Mouse Model of Homocystinuria
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

CBS, MET Act Sheet
Newborn Screening ACT Sheet Increased Methionine Homocystinuria (CBS Deficiency) / Hypermethioninemia (MET) Differential Diagnosis: Classical homocystinuria (cystathionine β-synthase (CBS) deficiency); hypermethioninemia (MET) due to methionine adenosyltransferase I/III MAT I/III deficiency; glycine n-methyltransferase GNMT deficiency; adenosylhomocysteine hydrolase deficiency; liver disease; hyperalimentation. Condition Description: Methionine from ingested protein is normally converted to homocysteine. In classical homocystinuria due to CBS deficiency, homocysteine cannot be converted to cystathionine. As a result, the concentration of homocysteine and its precursor, methionine, will become elevated. In MAT I/III deficiency and the other hypermethioninemias, methionine is increased in the absence of, or only with, a slightly increased level of homocysteine. You Should Take the Following IMMEDIATE Actions • Contact family to inform them of the newborn screening result and ascertain clinical status. • Consult with pediatric metabolic specialist. (See attached list.) • Evaluate the newborn with attention to liver disease and refer as appropriate. • Initiate confirmatory/diagnostic tests in consultation with metabolic specialist. • Initial testing: Plasma quantitative amino acids and plasma total homocysteine. • Repeat newborn screen if second screen has not been done. • Educate family about homocystinuria and its management as appropriate. • Report findings to newborn screening program. Diagnostic Evaluation: Plasma quantitative amino -

Incidence of Inborn Errors of Metabolism by Expanded Newborn
Original Article Journal of Inborn Errors of Metabolism & Screening 2016, Volume 4: 1–8 Incidence of Inborn Errors of Metabolism ª The Author(s) 2016 DOI: 10.1177/2326409816669027 by Expanded Newborn Screening iem.sagepub.com in a Mexican Hospital Consuelo Cantu´-Reyna, MD1,2, Luis Manuel Zepeda, MD1,2, Rene´ Montemayor, MD3, Santiago Benavides, MD3, Hector´ Javier Gonza´lez, MD3, Mercedes Va´zquez-Cantu´,BS1,4, and Hector´ Cruz-Camino, BS1,5 Abstract Newborn screening for the detection of inborn errors of metabolism (IEM), endocrinopathies, hemoglobinopathies, and other disorders is a public health initiative aimed at identifying specific diseases in a timely manner. Mexico initiated newborn screening in 1973, but the national incidence of this group of diseases is unknown or uncertain due to the lack of large sample sizes of expanded newborn screening (ENS) programs and lack of related publications. The incidence of a specific group of IEM, endocrinopathies, hemoglobinopathies, and other disorders in newborns was obtained from a Mexican hospital. These newborns were part of a comprehensive ENS program at Ginequito (a private hospital in Mexico), from January 2012 to August 2014. The retrospective study included the examination of 10 000 newborns’ results obtained from the ENS program (comprising the possible detection of more than 50 screened disorders). The findings were the following: 34 newborns were confirmed with an IEM, endocrinopathies, hemoglobinopathies, or other disorders and 68 were identified as carriers. Consequently, the estimated global incidence for those disorders was 3.4 in 1000 newborns; and the carrier prevalence was 6.8 in 1000. Moreover, a 0.04% false-positive rate was unveiled as soon as diagnostic testing revealed negative results. -

Birth Prevalence of Disorders Detectable Through Newborn Screening by Race/Ethnicity
©American College of Medical Genetics and Genomics ORIGINAL RESEARCH ARTICLE Birth prevalence of disorders detectable through newborn screening by race/ethnicity Lisa Feuchtbaum, DrPH, MPH1, Jennifer Carter, MPH2, Sunaina Dowray, MPH2, Robert J. Currier, PhD1 and Fred Lorey, PhD1 Purpose: The purpose of this study was to describe the birth prev- Conclusion: The California newborn screening data offer a alence of genetic disorders among different racial/ethnic groups unique opportunity to explore the birth prevalence of many through population-based newborn screening data. genetic dis orders across a wide spectrum of racial/ethnicity classifications. The data demonstrate that racial/ethnic subgroups Methods: Between 7 July 2005 and 6 July 2010 newborns in Cali- of the California newborn population have very different patterns fornia were screened for selected metabolic, endocrine, hemoglobin, of heritable disease expression. Determining the birth prevalence and cystic fibrosis disorders using a blood sample collected via heel of these disorders in California is a first step to understanding stick. The race and ethnicity of each newborn was self-reported by the short- and long-term medical and treatment needs faced by the mother at the time of specimen collection. affected communities, especially those groups that are impacted by Results: Of 2,282,138 newborns screened, the overall disorder detec- more severe disorders. tion rate was 1 in 500 births. The disorder with the highest prevalence Genet Med 2012:14(11):937–945 among all groups was primary congenital hypothyroidism (1 in 1,706 births). Birth prevalence for specific disorders varied widely among Key Words: birth prevalence; disorders; newborn screening; race different racial/ethnic groups. -

Genes Investigated
BabyNEXTTM EXTENDED Investigated genes and associated diseases Gene Disease OMIM OMIM Condition RUSP gene Disease ABCC8 Familial hyperinsulinism 600509 256450 Metabolic disorder - ABCC8-related Inborn error of amino acid metabolism ABCD1 Adrenoleukodystrophy 300371 300100 Miscellaneous RUSP multisystem (C) * diseases ABCD4 Methylmalonic aciduria and 603214 614857 Metabolic disorder - homocystinuria, cblJ type Inborn error of amino acid metabolism ACAD8 Isobutyryl-CoA 604773 611283 Metabolic Disorder - RUSP dehydrogenase deficiency Inborn error of (S) ** organic acid metabolism ACAD9 acyl-CoA dehydrogenase-9 611103 611126 Metabolic Disorder - (ACAD9) deficiency Inborn error of fatty acid metabolism ACADM Acyl-CoA dehydrogenase, 607008 201450 Metabolic Disorder - RUSP medium chain, deficiency of Inborn error of fatty (C) acid metabolism ACADS Acyl-CoA dehydrogenase, 606885 201470 Metabolic Disorder - RUSP short-chain, deficiency of Inborn error of fatty (S) acid metabolism ACADSB 2-methylbutyrylglycinuria 600301 610006 Metabolic Disorder - RUSP Inborn error of (S) organic acid metabolism ACADVL very long-chain acyl-CoA 609575 201475 Metabolic Disorder - RUSP dehydrogenase deficiency Inborn error of fatty (C) acid metabolism ACAT1 Alpha-methylacetoacetic 607809 203750 Metabolic Disorder - RUSP aciduria Inborn error of (C) organic acid metabolism ACSF3 Combined malonic and 614245 614265 Metabolic Disorder - methylmalonic aciduria Inborn error of organic acid metabolism 1 ADA Severe combined 608958 102700 Primary RUSP immunodeficiency due -

Amino Acid Disorders
471 Review Article on Inborn Errors of Metabolism Page 1 of 10 Amino acid disorders Ermal Aliu1, Shibani Kanungo2, Georgianne L. Arnold1 1Children’s Hospital of Pittsburgh, University of Pittsburgh School of Medicine, Pittsburgh, PA, USA; 2Western Michigan University Homer Stryker MD School of Medicine, Kalamazoo, MI, USA Contributions: (I) Conception and design: S Kanungo, GL Arnold; (II) Administrative support: S Kanungo; (III) Provision of study materials or patients: None; (IV) Collection and assembly of data: E Aliu, GL Arnold; (V) Data analysis and interpretation: None; (VI) Manuscript writing: All authors; (VII) Final approval of manuscript: All authors. Correspondence to: Georgianne L. Arnold, MD. UPMC Children’s Hospital of Pittsburgh, 4401 Penn Avenue, Suite 1200, Pittsburgh, PA 15224, USA. Email: [email protected]. Abstract: Amino acids serve as key building blocks and as an energy source for cell repair, survival, regeneration and growth. Each amino acid has an amino group, a carboxylic acid, and a unique carbon structure. Human utilize 21 different amino acids; most of these can be synthesized endogenously, but 9 are “essential” in that they must be ingested in the diet. In addition to their role as building blocks of protein, amino acids are key energy source (ketogenic, glucogenic or both), are building blocks of Kreb’s (aka TCA) cycle intermediates and other metabolites, and recycled as needed. A metabolic defect in the metabolism of tyrosine (homogentisic acid oxidase deficiency) historically defined Archibald Garrod as key architect in linking biochemistry, genetics and medicine and creation of the term ‘Inborn Error of Metabolism’ (IEM). The key concept of a single gene defect leading to a single enzyme dysfunction, leading to “intoxication” with a precursor in the metabolic pathway was vital to linking genetics and metabolic disorders and developing screening and treatment approaches as described in other chapters in this issue. -

Formula Name Category Description Qualifying
Memorandum #17-079 TO: WIC Regional Directors WIC Local Agency Directors FROM: Amanda Hovis, Director Nutrition Education/Clinic Services Unit Nutrition Services Section DATE: August 4, 2017 SUBJECT: Revised Formula Approval Resources Posted The formula approval resources have been revised to reflect the recent clinic formula table changes and will be posted to the DSHS WIC website. You will be able view them at the following link under “Formula Approval Resources” once they are posted. http://www.dshs.texas.gov/wichd/nut/foods-nut.shtm The following documents have been revised for July, 2017. Presently, they are attached to this memo as PDF files. 1. Texas WIC Formulary 2. Formula Code List 3. Texas WIC Formula Maximum Quantity Table 4. Nutrition Assessment Requirements Guide If you have questions or require additional information, contact Pat Koym, Formula Specialist, at [email protected] or 512-341-4578. This institution is an equal opportunity provider TEXAS WIC FORMULARY AND MEDICAL REASONS FOR ISSUANCE JULY 2017 Formula Category Description Qualifying Conditions Staff Instructions - May issue for 1 cert Manufacturer Name period unless otherwise indicated Alfamino Infant Elemental 20 cal/oz when mixed 1 scoop to 1 oz 1) Malabsorption syndrome Formula history required. Nestle water; hypoallergenic amino acid 2) GI impairment When requested for food allergy - a failed trial of a protein based elemental. 43% of fat is MCT 3) GER/GERD hydrolysate (Extensive HA, Nutramigen, Alimentum, or oil; Similar to Elecare DHA/ARA, 4) Food allergies (cow's milk, soy or Pregestimil) is recommended before issuing unless medically Neocate DHA/ARA and PurAmino. -

Living with Classical Homocystinuria
Living with Classical Homocystinuria This brochure will help you understand what classical homocystinuria is, how it affects your body, and how you can manage your condition A few words about this brochure What is homocystinuria? Has your doctor diagnosed you or your child You may have heard the word “homocystinuria” with homocystinuria (HO-mo-SIS-tin-YUR- for the first time when your doctor talked to ee-uh)? There are three types of genetic you about possibly having this condition. disorders that cause homocystinuria. Each Homocystinuria is a rare disorder involving type has a different cause and different the amino acid homocysteine (HO-mo-SIS- health issues. This brochure will talk about teen). Amino acids are building blocks that your classical homocystinuria. The information body uses to make proteins. Homocystinuria will help you understand classical occurs when there is a buildup of the amino acid homocystinuria and how you can manage homocysteine in your blood and urine. your condition. High levels of homocysteine can be harmful to your body. You may be reading this brochure because you have classical homocystinuria or Why is there homocysteine because your child or a sibling or a friend in your body? has it. Or perhaps you’re a healthcare professional. Please note the brochure It starts with the foods you eat. Your body addresses “you,” but it’s understood that makes homocysteine from another amino acid “you,” the reader, may not have classical called methionine (meh-THIGH-uh-neen). Most homocystinuria yourself. foods contain some methionine. But high-protein foods such as meat, fish, eggs, or cheese tend to have the most methionine. -

Web-Based Newborn Screening System for Metabolic Diseases: Machine Learning Versus Clinicians
JOURNAL OF MEDICAL INTERNET RESEARCH Chen et al Original Paper Web-Based Newborn Screening System for Metabolic Diseases: Machine Learning Versus Clinicians Wei-Hsin Chen1, MS; Sheau-Ling Hsieh2, PhD; Kai-Ping Hsu3, PhD; Han-Ping Chen4, BS; Xing-Yu Su1, BS; Yi-Ju Tseng1, BS; Yin-Hsiu Chien5, MD, PhD; Wuh-Liang Hwu5, MD, PhD; Feipei Lai1,4,6, PhD 1National Taiwan University, Graduate Institute of Biomedical Electronics and Bioinformatics, Taipei, Taiwan 2National Chiao Tung University, Hsinchu, Taiwan 3National Taiwan University, Computer and Information Networking Center, Taipei, Taiwan 4National Taiwan University, Department of Computer Science and Information Engineering, Taipei, Taiwan 5National Taiwan University Hospital, Department of Medical Genetics, Taipei, Taiwan 6National Taiwan University, Department of Electrical Engineering, Taipei, Taiwan Corresponding Author: Sheau-Ling Hsieh, PhD National Chiao Tung University 1001, University Road, Hsinchu, Taiwan Hsinchu, 30010 Taiwan Phone: 886 3 513 1351 Fax: 886 3 571 4031 Email: [email protected] Abstract Background: A hospital information system (HIS) that integrates screening data and interpretation of the data is routinely requested by hospitals and parents. However, the accuracy of disease classification may be low because of the disease characteristics and the analytes used for classification. Objective: The objective of this study is to describe a system that enhanced the neonatal screening system of the Newborn Screening Center at the National Taiwan University Hospital. The system was designed and deployed according to a service-oriented architecture (SOA) framework under the Web services .NET environment. The system consists of sample collection, testing, diagnosis, evaluation, treatment, and follow-up services among collaborating hospitals. -

Suggested Follow-Up for Homocystinuria Elevated Methionine (MET)
Suggested Follow-up for Homocystinuria Elevated Methionine (MET) Possible Causes: Elevated methionine (MET) is the primary marker for homocystinuria. This disorder is primarily caused by a deficiency in the enzyme cystothionine synthetase. Untreated infants are at risk for developmental delay, mental retardation, dislocated ocular lens, myopia and thromboembolism. Screening for homocystinuria may also identify infants with hypermethioninemia. Primary hypermethioninemia that is not caused by other disorders, liver disease or excess methionine intake appears to be extremely rare. Next Steps if Abnormal: See infant as soon as possible to ascertain health status. Consult pediatric metabolic specialist and initiate diagnostic evaluation and treatment as recommended. Common diagnostic studies include plasma amino acids, total plasma homocysteine and urine amino acids. In addition, repeat amino acid profile on filter paper and send to the DHEC laboratory. Neonatal Presentation: Usually none. Emergency Treatment: Usually none necessary. Standard Treatment: MET restricted diet for life. Some affected persons are responsive to Vitamin B6 and may not need MET restricted diet. Betaine often used. Advice for Family: Provide basic information about homocystinuria. The handout, When Baby Needs a Second Test for Homocystinuria, may be used for this purpose. Internet Resources: http://oregon.gov/DHS/ph/nbs/expand.shtml http://web1.tch.harvard.edu/newenglandconsortium/scientists_physicians2.html http://ghr.nlm.nih.gov/condition=homocystinuria http://ghr.nlm.nih.gov/condition=hypermethioninemia http://www.acmg.net/resources/policies/ACT/condition-analyte-links.htm . -
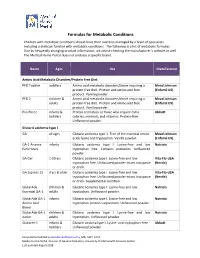
Formulas for Metabolic Conditions
Formulas for Metabolic Conditions Children with metabolic conditions should have their nutrition managed by a team of specialists including a dietician familiar with metabolic conditions. The following is a list of metabolic formulas. Due to frequently changing product information, we advise checking the manufacturer’s website as well. The Medical Home Portal does not endorse a specific brand. Name Ages Use Manufacturer Amino Acid Metabolic Disorders/Protein Free Diet PFD Toddler toddlers Amino acid metabolic disorders/those requiring a Mead Johnson protein‐free diet. Protein and amino acid free (Enfamil US) product. Vanilla powder. PFD 2 children & Amino acid metabolic disorders/those requiring a Mead Johnson adults protein‐free diet. Protein and amino acid free (Enfamil US) product. Vanilla powder. Pro‐Phree infants & Protein restriction or those who require extra Abbott toddlers calories, minerals, and vitamins. Protein‐free. Unflavored powder. Glutaric acidemia type 1 GA all ages Glutaric acidemia type 1. Free of the essential amino Mead Johnson acids lysine and tryptophan. Vanilla powder. (Enfamil US) GA‐1 Anamix infants Glutaric acidemia type I. Lysine‐free and low Nutricia Early Years tryptophan free. Contains prebiotics. Unflavored powder. GA Gel 1‐10 yrs Glutaric acidemia type I. Lysine‐free and low Vita‐Flo‐USA tryptophan free. Unflavored powder mixes into paste (Nestle) or drink. GA Express 15 3 yrs & older Glutaric acidemia type I. Lysine‐free and low Vita‐Flo‐USA tryptophan free. Unflavored powder mixes into paste (Nestle) or drink. Supplemental nutrition. GlutarAde children & Glutaric acidemia type I. Lysine‐free and low Nutricia Essential GA‐1 adults tryptophan. Unflavored powder. -

Texas Newborn Screening Panel
TEXAS NEWBORN SCREENING PANEL BLOODSPOT TESTING (conducted at DSHS Laboratory) Amino Acid Disorders Core Conditions Secondary Conditions • Argininosuccinic Aciduria (ASA) • Argininemia (ARG) • Citrullinemia, Type I (CIT) • Benign Hyperphenylalaninemia (H-PHE) • Homocystinuria (HCY) • Biopterin defect in cofactor biosynthesis (BIOPT BS) • Maple Syrup Urine Disease (MSUD) • Biopterin defect in cofactor regeneration (BIOPT REG) • Classic Phenylketonuria (PKU) • Citrullinemia, Type II (CIT II) • Tyrosinemia, Type I (TYR I) • Hypermethioninemia (MET) • Tyrosinemia, Type II (TYR II) • Tyrosinemia, Type III (TYR III) Fatty Acid Disorders Core Conditions Secondary Conditions • Carnitine Uptake Defect (CUD) • 2,4 Dienoyl-CoA Reductase Deficiency (DE RED) • Long Chain L-3-Hydroxyacyl-CoA Dehydrogenase Deficiency • Carnitine Acylcarnitine Translocase Deficiency (CACT) (LCHAD) • Carnitine Palmitoyltransferase Type I Deficiency (CPT I) • Medium-Chain Acyl-CoA Dehydrogenase Deficiency (MCAD) • Carnitine Palmitoyltransferase Type II Deficiency (CPT II) • Trifunctional Protein Deficiency (TFP) • Glutaric Acidemia Type II (GA2) • Very Long-Chain Acyl-CoA Dehydrogenase Deficiency (VLCAD) • Medium-Chain Ketoacyl-CoA Thiolase Deficiency (MCKAT) • Medium/Short Chain L-3-Hydroxyacyl-CoA Dehydrogenase Deficiency (M/SCHAD) • Short-Chain Acyl-CoA Dehydrogenase Deficiency (SCAD) Organic Acid Disorders Core Conditions Secondary Conditions • 3-Methylcrotonyl-CoA Carboxylase Deficiency (3-MCC) • 2 Methylbutyrylglycinuria (2MBG) • 3-Hydroxy-3-Methylglutaric Aciduria -
![Newborn Screening ACT Sheet [Increased Methionine] Homocystinuria (CBS Deficiency)](https://docslib.b-cdn.net/cover/6249/newborn-screening-act-sheet-increased-methionine-homocystinuria-cbs-deficiency-2586249.webp)
Newborn Screening ACT Sheet [Increased Methionine] Homocystinuria (CBS Deficiency)
American College of Medical Genetics ACT SHEET Newborn Screening ACT Sheet [Increased Methionine] Homocystinuria (CBS Deficiency) Differential Diagnosis: Classical homocystinuria (cystathionine ß-synthase (CBS) deficiency); hypermethioninemia due to methionine adenosyltransferase I/III (MAT I/III) deficiency; glycine n-methyltransferase (GNMT) deficiency; adenosylhomocysteine hydrolase deficiency; liver disease; hyperalimentation. Condition Description: Methionine from ingested protein is normally converted to homocysteine. In classical homocystinuria due to CBS deficiency, homocysteine cannot be converted to cystathionine. As a result, the concentration of homocysteine and its precursor, methionine, will become elevated. In MAT I/III deficiency and the other hypermethioninemias, methionine is increased in the absence of or only with a slightly increased level of homocysteine. YOU SHOULD TAKE THE FOLLOWING ACTIONS: . Contact family to inform them of the newborn screening result and ascertain clinical status. Consult with pediatric metabolic specialist. Evaluate the newborn with attention to liver disease and refer as appropriate. Initiate confirmatory/diagnostic tests in consultation with metabolic specialist. Educate family about homocystinuria and its management, as appropriate. Report findings to newborn screening program. Diagnostic Evaluation: Quantitative plasma amino acids will show increased homocystine and methionine in classical homocystinuria but only increased methionine in the other disorders. Plasma homocysteine analysis will show markedly increased homocysteine in classical homocystinuria and normal or only slightly increased homocysteine in the other disorders. Urine homocysteine is markedly increased in classical homocystinuria. Clinical Considerations: Homocystinuria is usually asymptomatic in the neonate. If untreated, these children eventually develop mental retardation, ectopia lentis, a marfanoid appearance including arachnodactyly, osteoporosis, other skeletal deformities and thromboembolism. MAT I/III deficiency may be benign.