C9orf72 Frontotemporal Dementia and Amyotrophic Lateral Sclerosis: Investigating Repeat Pathology in Cell Culture Models and Human Post-Mortem Brain
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Greg J Sheehan, FWS Principal Deputy Director
Greg J Sheehan, FWS Principal Deputy Director Tue Jun 6, 2017 2:45pm FWS weekly check in meeting Video call: (b) (5), (b) (6) Where: AS/FWP Conference Room -- 3144 Calendar: Maureen Foster Created by: Tasha Robbins Who: Thomas Irwin, Charisa Morris, Maureen Foster, Richard Goeken, GregSheehan, Wendy Fink, Stephen Guertin, Catherine Gulac, Jason Larrabee,Tasha Robbins, Aurelia Skipwith, Marshall Critchfield, Heather Swift,Jim Kurth, Todd Willens, Roslyn Sellars, Zachariah Gambill, PegRomanik, Barbara Wainman 3pm *FWS weekly check in meeting Video call: (b) (5), (b) (6) Where: AS/FWP Conference Room -- 3144 Calendar: Greg Sheehan Created by: Roslyn Sellars Wed Jun 7, 2017 1pm OIG monthly status update meeting/conference call (Jim, Steve, Kathy Garrity, Keith Toomey, Charisa) Room 3357 Video call: (b) (5), (b) (6) Where: Dial: (b) (5) , Code: (b) (5) Calendar: Jim Kurth Created by: Roslyn Sellars Who: Casey Hammond, Stephen Guertin, Katherine Garrity, Jim Kurth, GregSheehan, Charisa Morris, Keith Toomey 2:30pm [Asst Directors/Regional Directors Only-No Deputies or Actings] - Weekly Directorate VTC: Transition Check-In--Room 3038 Video call: (b) (5), (b) (6) Calendar: Jim Kurth Created by: Thomas Irwin Who: [email protected], Tom Melius, [email protected], WandaCantrell, Edward Grace, [email protected], Henry Schlitzer,Pamela Michalegko, Kenneth Taylor, Benjamin Tuggle, Brian Bloodsworth,Robyn Thorson, Gary Frazer, Michael Gale, Jim Kurth, Charisa Morris,Paul Rauch, Denise Thompson, Seth Mott, Cynthia Martinez,cynthia_dohner@fws -
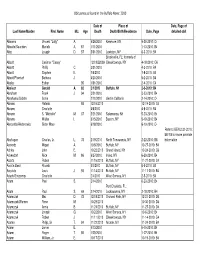
Obituaries Buffalo News 2010 by Name
Obituaries as found in the Buffalo News: 2010 Date of Place of Date, Page of Last Name/Maiden First Name M.I. Age Death Death/Birth/Residence Date, Page detailed obit Abbarno Vincent "Lolly" A. 9/26/2010 Kenmore, NY 9-30-2010: C4 Abbatte/Saunders Murielle A. 87 1/11/2010 1-13-2010: B4 Abbo Joseph D. 57 5/31/2010 Lewiston, NY 6-3-2010: B4 Brooksville, FL; formerly of Abbott Casimer "Casey" 12/19/22009 Cheektowaga, NY 4-18-2010: C6 Abbott Phillip C. 3/31/2010 4-3-2010: B4 Abbott Stephen E. 7/6/2010 7-8-2010: B4 Abbott/Pfoetsch Barbara J. 4/20/2010 5-2-2010: B4 Abeles Esther 95 1/31/2010 2-4-2010: C4 Abelson Gerald A. 82 2/1/2010 Buffalo, NY 2-3-2010: B4 Abraham Frank J. 94 3/21/2010 3-23-2010: B4 Abrahams/Gichtin Sonia 2/10/2010 died in California 2-14-2010: C4 Abramo Rafeala 93 12/16/2010 12-19-2010: C4 Abrams Charlotte 4/6/2010 4-8-2010: B4 Abrams S. "Michelle" M. 37 5/21/2010 Salamanca, NY 5-23-2010: B4 Abrams Walter I. 5/15/2010 Basom, NY 5-19-2010: B4 Abrosette/Aksterowicz Sister Mary 6/18/2010 6-19-2010: C4 Refer to BEN 2-21-2010: B6/7/8 for more possible Abshagen Charles, Jr. L. 73 2/19/2010 North Tonawanda, NY 2-22-2010: B8 information Acevedo Miguel A. 10/6/2010 Buffalo, NY 10-27-2010: B4 Achkar John E. -

Vol. 83 Wednesday, No. 211 October 31, 2018 Pages 54663–54860
Vol. 83 Wednesday, No. 211 October 31, 2018 Pages 54663–54860 OFFICE OF THE FEDERAL REGISTER VerDate Sep 11 2014 20:07 Oct 30, 2018 Jkt 247001 PO 00000 Frm 00001 Fmt 4710 Sfmt 4710 E:\FR\FM\31OCWS.LOC 31OCWS amozie on DSK3GDR082PROD with FRONT MATTER WS II Federal Register / Vol. 83, No. 211 / Wednesday, October 31, 2018 The FEDERAL REGISTER (ISSN 0097–6326) is published daily, SUBSCRIPTIONS AND COPIES Monday through Friday, except official holidays, by the Office PUBLIC of the Federal Register, National Archives and Records Administration, under the Federal Register Act (44 U.S.C. Ch. 15) Subscriptions: and the regulations of the Administrative Committee of the Federal Paper or fiche 202–512–1800 Register (1 CFR Ch. I). The Superintendent of Documents, U.S. Assistance with public subscriptions 202–512–1806 Government Publishing Office, is the exclusive distributor of the official edition. Periodicals postage is paid at Washington, DC. General online information 202–512–1530; 1–888–293–6498 Single copies/back copies: The FEDERAL REGISTER provides a uniform system for making available to the public regulations and legal notices issued by Paper or fiche 202–512–1800 Federal agencies. These include Presidential proclamations and Assistance with public single copies 1–866–512–1800 Executive Orders, Federal agency documents having general (Toll-Free) applicability and legal effect, documents required to be published FEDERAL AGENCIES by act of Congress, and other Federal agency documents of public Subscriptions: interest. Assistance with Federal agency subscriptions: Documents are on file for public inspection in the Office of the Federal Register the day before they are published, unless the Email [email protected] issuing agency requests earlier filing. -

Magazin Wirtschaft
www.stuttgart.ihk.de 07.2013 Stuttgart - Böblingen - Esslingen-Nürtingen - Göppingen - Ludwigsburg - Rems-Murr Magazin Wirtschaft Ein Service der IHK für Unternehmen in der Region Stuttgart Fachkräfte finden zwischen Alb und Gäu Seite 6 wer hat schon ein ganzes kabinett nur für diewirtschaft? Selbstverständlich wir. Damit die Betriebe gute Rahmenbedingungen haben, sind in unseren Gremien ausschließlich Wirtschaftexperten vertreten. www.stuttgart.ihk.de oder Infoline 0711 2005-0 EDITORIAL Handelskriege kennen keine Gewinner Ähnlich dem Frühjahrswetter in diesem weit kommen? China ist die zweitgrößte Wirt- Jahr ziehen nun auch Wolken über dem schaftsmacht der Welt. Daher sollte das Land Parkett des internationalen Handels beginnen, sich an internationale Spielregeln auf. Auslöser ist die vorläufige Erhebung von zu halten. Globalisierung benötigt faire Wett- EU-Strafzöllen auf chinesische Solarmodule. bewerbsbedingungen, an die sich China zu- China kontert mit einem Anti-Dumping-Ver- weilen jedoch nicht hält. Belegte Fälle von fahren gegen europäischen Wein. Immer Wettbewerbsverstößen gibt es genug. mehr Produkte treten in den Fokus der Dum- ping-Diskussion. Vernünftige Handelspolitik Die EU muss schnellstens sieht anders aus. Mangels Deeskalation wird Verhandlungen aufnehmen die Situation für China und Europa immer tassilo zywietz unangenehmer. Ein Handelskrieg droht. Europa darf davor die Augen nicht ver- Geschäftsführer International Globalisierung erfordert jedoch gegensei- schließen. Nachgiebigkeit ist keine Erfolg ver- der IHK Region Stuttgart tige Rücksichtnahme und Handeln im Sinne sprechende Strategie – damit würde die EU der immer stärker miteinander verbundenen erpressbar. Lässt man China gewähren, kann Volkswirtschaften: China und die EU haben die Methode künftig in der Chemiebranche letztes Jahr Waren und Dienstleistungen in und irgendwann im Autobau angewendet Höhe von über 400 Milliarden Euro ausge- werden. -

A-To-I RNA Editing Does Not Change with Age in the Healthy Male Rat Brain
Biogerontology (2013) 14:395–400 DOI 10.1007/s10522-013-9433-8 RESEARCH ARTICLE A-to-I RNA editing does not change with age in the healthy male rat brain Andrew P. Holmes • Shona H. Wood • Brian J. Merry • Joa˜o Pedro de Magalha˜es Received: 18 January 2013 / Accepted: 15 May 2013 / Published online: 26 May 2013 Ó The Author(s) 2013. This article is published with open access at Springerlink.com Abstract RNA editing is a post-transcriptional pro- Introduction cess, which results in base substitution modifications to RNA. It is an important process in generating Adenosine to inosine (A-to-I) RNA editing is a post- protein diversity through amino acid substitution and transcriptional process that alters the sequences of the modulation of splicing events. Previous studies RNA molecules. The adenosine deaminases ADAR have suggested a link between gene-specific reduc- and ADARB1 convert specific adenosine residues on tions in adenosine to inosine RNA editing and aging in RNA to inosine bases. During translation, sequencing, the human brain. Here we demonstrate that changes in and splicing, inosine is recognized as guanosine. RNA editing observed in humans with age are not Therefore, A-to-I RNA editing has important impli- observed during aging in healthy rats. Furthermore, we cations in altering specific amino acids, miRNA identify a conserved editing site in rats, in Cog3.We targeting, and in the modulation of alternative splicing propose that either age-related changes in RNA (Nishikura 2010). editing are specific to primates or humans, or that Targets of A-to-I RNA editing are often genes they are the manifestation of disease pathology. -

2013 Kaiser Permanente-Authored Publications Alphabetical by Author
2013 Kaiser Permanente-Authored Publications Alphabetical by Author 1. Abbas MA, Cannom RR, Chiu VY, Burchette RJ, Radner GW, Haigh PI, Etzioni DA Triage of patients with acute diverticulitis: are some inpatients candidates for outpatient treatment? Colorectal Dis. 2013 Apr;15(4):451-7. Southern California 23061533 KP author(s): Abbas, Maher A; Chiu, Vicki Y; Burchette, Raoul J; Radner, Gary W; Haigh, Philip I 2. Abbass MA, Slezak JM, DiFronzo LA Predictors of Early Postoperative Outcomes in 375 Consecutive Hepatectomies: A Single- institution Experience Am Surg. 2013 Oct;79(10):961-7. Southern California 24160779 KP author(s): Abbass, Mohammad Ali; Slezak, Jeffrey M; DiFronzo, Andrew L 3. Abbenhardt C, Poole EM, Kulmacz RJ, Xiao L, Curtin K, Galbraith RL, Duggan D, Hsu L, Makar KW, Caan BJ, Koepl L, Owen RW, Scherer D, Carlson CS, Genetics and Epidemiology of Colorectal Cancer Consortium (GECCO) and CCFR, Potter JD, Slattery ML, Ulrich CM Phospholipase A2G1B polymorphisms and risk of colorectal neoplasia Int J Mol Epidemiol Genet. 2013 Sep 12;4(3):140-9. Northern California 24046806 KP author(s): Caan, Bette 4. Aberg JA, Gallant JE, Ghanem KG, Emmanuel P, Zingman BS, Horberg MA Primary Care Guidelines for the Management of Persons Infected With HIV: 2013 Update by the HIV Medicine Association of the Infectious Diseases Society of America Clin Infect Dis. 2013 Nov 13. Mid-Atlantic 24235263 KP author(s): Horberg, Michael 5. Abraham AG, D'Souza G, Jing Y, Gange SJ, Sterling TR, Silverberg MJ, Saag MS, Rourke SB, Rachlis A, Napravnik S, Moore RD, Klein MB, Kitahata MM, Kirk GD, Hogg RS, Hessol NA, Goedert JJ, Gill MJ, Gebo KA, Eron JJ, Engels EA, Dubrow R, Crane HM, Brooks JT, Bosch RJ, Strickler HD, North American AIDS Cohort Collaboration on Research and Design of IeDEA Invasive cervical cancer risk among HIV-infected women: A North American multi-cohort collaboration prospective study J Acquir Immune Defic Syndr. -

Elecciones Anuales Grado 2020 − Delegados De Curso Página 1 De 51
Elecciones anuales Grado 2020 − Delegados de curso Fecha de impresión: 27−11−2020 Grado en Ingeniería Eléctrica − Curso: 1 Nº Apellido1 Apellido2 Nombre DNI 1 ARIAS RODRIGUEZ GABRIEL 7***03*3 2 CAMPOS LUCAS ALEJANDRO 7*18***9 3 FERNANDEZ MIERA ADRIAN *2***144 4 GARCÍA GARCÍA IÑAKI 20*0*3** 5 GARCIA HERRANZ JULIO *2*94**8 6 GONZALEZ VENERO HERNAN **08*08* 7 GUERRA SAINZ FRANCISCO RAMON **104**6 8 MADARIAGA GONZALEZ ERNESTO 7*103*** 9 MARCANO CUBILLAS MANUEL 7**2*0*1 10 MAZO LOPEZ PEDRO PABLO DEL **0*26*0 11 MOYA MANTECON LAURA 7*1**15* 12 ORTIZ SAMPERIO VICTOR *21***11 13 PEREDA SOLARES JORGE **1*501* 14 RAMOS MARTINEZ SERGIO 7**8**77 15 RODRIGUEZ EIZAGUIRRE JORGE 7*0**1*8 16 ROJAS LOPEZ FELIPE ***00*03 17 SOLANO RUIZ ALVARO ***0319* 18 XIANG QIXIN ****6291 En cumplimiento de lo dispuesto apartado 1 de la Disposición adicional séptima de la Ley Orgánica 3/2018, de 5 de diciembre, de Protección de Datos Personales y garantía de los derechos digitales, las personas que figuran en este listado aparecen identificadas con cuatro cifras numéricas aleatorias del DNI, NIE, Pasaporte o documento equivalente. En los casos en que el número de estos documentos tenga menos de seis cifras numéricas, se asignará un número aleatorio confeccionado con este fin. Los números aleatorios asignados pueden consultarse en Campus Virtual (Menú general / Datos personales). Página 1 de 51 Elecciones anuales Grado 2020 − Delegados de curso Fecha de impresión: 27−11−2020 Grado en Ingeniería Eléctrica − Curso: 2 Nº Apellido1 Apellido2 Nombre DNI 1 ALONSO MURGA RAUL -

Family Group Sheets Surname Index
PASSAIC COUNTY HISTORICAL SOCIETY FAMILY GROUP SHEETS SURNAME INDEX This collection of 660 folders contains over 50,000 family group sheets of families that resided in Passaic and Bergen Counties. These sheets were prepared by volunteers using the Societies various collections of church, ceme tery and bible records as well as city directo ries, county history books, newspaper abstracts and the Mattie Bowman manuscript collection. Example of a typical Family Group Sheet from the collection. PASSAIC COUNTY HISTORICAL SOCIETY FAMILY GROUP SHEETS — SURNAME INDEX A Aldous Anderson Arndt Aartse Aldrich Anderton Arnot Abbott Alenson Andolina Aronsohn Abeel Alesbrook Andreasen Arquhart Abel Alesso Andrews Arrayo Aber Alexander Andriesse (see Anderson) Arrowsmith Abers Alexandra Andruss Arthur Abildgaard Alfano Angell Arthurs Abraham Alje (see Alyea) Anger Aruesman Abrams Aljea (see Alyea) Angland Asbell Abrash Alji (see Alyea) Angle Ash Ack Allabough Anglehart Ashbee Acker Allee Anglin Ashbey Ackerman Allen Angotti Ashe Ackerson Allenan Angus Ashfield Ackert Aller Annan Ashley Acton Allerman Anners Ashman Adair Allibone Anness Ashton Adams Alliegro Annin Ashworth Adamson Allington Anson Asper Adcroft Alliot Anthony Aspinwall Addy Allison Anton Astin Adelman Allman Antoniou Astley Adolf Allmen Apel Astwood Adrian Allyton Appel Atchison Aesben Almgren Apple Ateroft Agar Almond Applebee Atha Ager Alois Applegate Atherly Agnew Alpart Appleton Atherson Ahnert Alper Apsley Atherton Aiken Alsheimer Arbuthnot Atkins Aikman Alterman Archbold Atkinson Aimone -

Structural Studies of C9orf72-SMCR8-WDR41 Protein Complex
Structural Studies of C9orf72-SMCR8-WDR41 Protein Complex Valeria Shkuratova Department of Biochemistry McGill University, Montreal A thesis submitted to McGill University in partial fulfillment of the requirements of the degree of Master of Science © Valeria Shkuratova, 2020 Table of Contents Abstract ............................................................................................................................................ 3 Résumé ............................................................................................................................................ 4 Acknowledgment ............................................................................................................................. 5 Author Contribution ........................................................................................................................ 6 List of Abbreviations ....................................................................................................................... 7 List of Figures .................................................................................................................................. 9 List of Tables ................................................................................................................................... 9 Introduction ................................................................................................................................... 10 1. Amyotrophic Lateral Sclerosis (ALS) .............................................................................. -

The Proline/Arginine Dipeptide from Hexanucleotide Repeat Expanded C9ORF72 Inhibits the Proteasome
New Research Disorders of the Nervous System The Proline/Arginine Dipeptide from Hexanucleotide Repeat Expanded C9ORF72 Inhibits the Proteasome Jelena Mojsilovic-Petrovic,4 Won Hoon Choi,5 Nathaniel Safren,6 ء,Matthews Lan,3 ء,Rahul Gupta,1,2 Sami Barmada,6 Min Jae Lee,5 and Robert Kalb4,7 DOI:http://dx.doi.org/10.1523/ENEURO.0249-16.2017 1Department of Chemical and Biomolecular Engineering, School of Engineering and Applied Sciences, University of Pennsylvania, Philadelphia, PA, 19104, 2Department of Biology, College of Arts and Sciences, University of Pennsylvania, Philadelphia, PA, 19104, 3Department of Biochemistry, College of Arts and Sciences, University of Pennsylvania, Philadelphia, PA, 19104, 4Division of Neurology, Department of Pediatrics, Research Institute, Children’s Hospital of Philadelphia, Philadelphia, PA, 19104, 5Department of Biochemistry and Molecular Biology, Seoul National University College of Medicine, Seoul 03080, Republic of Korea, 6Department of Neurology, University of Michigan, Ann Arbor, MI, 48109, and 7Department of Neurology, Perelman School of Medicine, University of Pennsylvania, Philadelphia, PA, 19104 Abstract An intronic hexanucleotide repeat expansion (HRE) mutation in the C9ORF72 gene is the most common cause of familial ALS and frontotemporal dementia (FTD) and is found in ϳ7% of individuals with apparently sporadic disease. Several different diamino acid peptides can be generated from the HRE by noncanonical translation (repeat-associated non-ATG translation, or RAN translation), and some of these peptides can be toxic. Here, we studied the effects of two arginine containing RAN translation products [proline/arginine repeated 20 times (PR20) and glycine/arginine repeated 20 times (GR20)] in primary rat spinal cord neuron cultures grown on an astrocyte feeder layer. -

C9orf72 Testing in a Diagnostic Laboratory Using the Asuragen Amplidex® PCR/CE C9orf72 Kit
C9orf72 testing in a diagnostic laboratory using the Asuragen AmplideX® PCR/CE C9orf72 kit Rick van Minkelen1, Patricia Reichgelt1, Saskia Theunisse2, Jody Salomon2, Kristen Culp3, Dept. Clinical Genetics Janne M. Papma4, Laura Donker Kaat4,5, John C. van Swieten4,6, Raoul van de Graaf1, Lies H. Hoefsloot1, Martina Wilke1 1 Department of Clinical Genetics, Erasmus Medical Center, Rotterdam, The Netherlands 2 Sanbio B.V., Uden, the Netherlands 3 Asuragen, Inc., Austin, TX, USA 4 Department of Neurology, Erasmus MC, Rotterdam, the Netherlands 5 Department of Clinical Genetics, LUMC, Leiden, the Netherlands 6 Department of Clinical Genetics, VUMC, Amsterdam, the Netherlands [email protected] Introduction The pathogenic expanded hexanucleotide repeat element (G4C2) in intron 1 of the Chromosome 9 open reading frame 72 gene (C9orf72; Results NM_001256054.2) is the most prevalent genetic cause of the All samples previously scored as pathogenic using the home-made tests neurodegenerative disorders frontotemporal dementia (FTD) and also showed a pathogenic repeat expansion of at least 145 repeats using amyotrophic lateral sclerosis (ALS). Here we describe our experiences in the new Asuragen test (for examples see Figure 1). No repeats were sized using the newly introduced Asuragen AmplideX® PCR/CE C9orf72 assay in the range 60-145. Repeats in the normal range were sized exactly the compared to our home-made long range and repeat-primed PCR tests for same. DNA concentrations of 50ng/µl (our standard lab dilution) and the detection of C9orf72 repeats. Our home-made tests are limited to 25ng/µl showed similar results. A dilution (including a second 30s injection detection of up to ~60 C9orf72 repeats. -

Altered Regulation of Adipomir Editing with Aging
International Journal of Molecular Sciences Article Altered Regulation of adipomiR Editing with Aging Sabel Meadows, Abbagael Seidler, Madison Wall, Jamika Page, Cara Taylor, Brendin Flinn , Robin Turner and Nalini Santanam * Department of Biomedical Sciences, Joan C Edwards School of Medicine, Marshall University, Huntington, WV 25755, USA; [email protected] (S.M.); [email protected] (A.S.); [email protected] (M.W.); [email protected] (J.P.); [email protected] (C.T.); fl[email protected] (B.F.); [email protected] (R.T.) * Correspondence: [email protected] Received: 18 August 2020; Accepted: 17 September 2020; Published: 20 September 2020 Abstract: Adipose dysfunction with aging increases risk to insulin resistance and other chronic metabolic diseases. We previously showed functional changes in microRNAs involved in pre-adipocyte differentiation with aging resulting in adipose dysfunction. However, the mechanisms leading to this dysfunction in microRNAs in adipose tissue (adipomiRs) during aging are not well understood. We determined the longitudinal changes in expression of adipomiRs and studied their regulatory mechanisms, such as miRNA biogenesis and editing, in an aging rodent model, with Fischer344 Brown-Norway hybrid rats at ages ranging from 3 to 30 months (male/females, × n > 8). Expression of adipomiRs and their edited forms were determined by small-RNA sequencing. RT-qPCR was used to measure the mRNA expression of biogenesis and editing enzymes. Sanger sequencing was used to validate editing with aging. Differential expression of adipomiRs involved in adipocyte differentiation and insulin signaling was altered with aging. Sex- and age-specific changes in edited adipomiRs were observed. An increase in miRNA biogenesis and editing enzymes (ADARs and their splice variants) were observed with increasing age, more so in female than male rats.