Phylogeography of the Common Pheasant Phasianus Colchinus
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Keeping Pheasants
KKKEEEEEEPPPIIINNNGGG PPPHHHEEEAAASSSAAANNNTTTSSS AAASSS AAA HHHOOOBBBBBBYYY Text and photos: Jan Willem Schrijvers Photo above: Siamese Fireback pheasant male(Lophura diardi). ORIGIN AND LIFESTYLE OF THE WILD PHEASANT Pheasants are wild gallinaceous birds all originating in Asia. One exception is the Congo peacock from Africa. (The pheasants also include game fowl and peacocks.) Each species has its own characteristics and life habits. There are species that live in tropical rain forests, but there are species that live in the mountains, on cold plains. This is something to take in account when housing our pheasants (with or without a night coop, with or without heating). Most species live in and around the mountains with a woodland vegetation, where they can find lots of berries, greens and seeds. It is of utmost importance to first consider the habits and living conditions of the species you want to keep. Only with proper housing will these beautiful birds show to their fullest, and reproduce. PHEASANTS IN AVIARIES In the past there were some "pheasant farms" which mainly bred the common pheasant (also known as ring-neck pheasant). The birds on these farms were bred for sport hunting. They were released en masse in the autumn in the fields and other areas to be shot for the sport. Some escaped from the hunters and that is why today we can see wild pheasants roam our fields and woods. These pheasants have also succeeded in reproducing themselves, even though they are not native birds. The birds are also kept for their colourful feathers. Each year I see Prince Carnival walk again with the beautiful feathers of the Reeves's Pheasant at his cocked hat. -

Conservation Genetics and Management of the Chukar Partridge Alectoris Chukar in Cyprus and the Middle East PANICOS PANAYIDES, MONICA GUERRINI & FILIPPO BARBANERA
Conservation genetics and management of the Chukar Partridge Alectoris chukar in Cyprus and the Middle East PANICOS PANAYIDES, MONICA GUERRINI & FILIPPO BARBANERA The Chukar Partridge Alectoris chukar (Phasianidae) is a popular game bird whose range extends from the Balkans to eastern Asia. The Chukar is threatened by human-mediated hybridization either with congeneric species (Red-Legged A. rufa and Rock A. graeca Partridges) from Europe or exotic conspecifics (from eastern Asia), mainly through introductions. We investigated Chukar populations of the Middle East (Cyprus, Turkey, Lebanon, Israel, Armenia, Georgia, Iran and Turkmenistan: n = 89 specimens) in order to obtain useful genetic information for the management of this species. We sequenced the entire mitochondrial DNA (mtDNA) Control Region using Mediterranean (Greece: n = 27) and eastern Asian (China: n = 18) populations as intraspecific outgroups. The Cypriot Chukars (wild and farmed birds) showed high diversity and only native genotypes; signatures of both demographic and spatial expansion were found. Our dataset suggests that Cyprus holds the most ancient A. chukar haplotype of the Middle East. We found A. rufa mtDNA lineage in Lebanese Chukars as well as A. chukar haplotypes of Chinese origin in Greek and Turkish Chukars. Given the very real risk of genetic pollution, we conclude that present management of game species such as the Chukar cannot avoid anymore the use of molecular tools. We recommend that Chukars must not be translocated from elsewhere to Cyprus. INTRODUCTION The distribution range of the most widespread species of Alectoris partridge, the Chukar (A. chukar, Phasianidae, Plate 1), is claimed to extend from the Balkans to eastern Asia. -

A Study of Food and Feeding Habits of Blue Peafowl, Pavo Cristatus Linnaeus, 1758 in District Kurukshetra, Haryana (India)
International Journal of Research Studies in Biosciences (IJRSB) Volume 2, Issue 6, July 2014, PP 11-16 ISSN 2349-0357 (Print) & ISSN 2349-0365 (Online) www.arcjournals.org A Study of Food and Feeding Habits of Blue Peafowl, Pavo Cristatus Linnaeus, 1758 in District Kurukshetra, Haryana (India) Girish Chopra, Tarsem Kumar Department of Zoology, Kurukshetra University, Kurukshetra-136119 (INDIA) [email protected] Summary: Present study was conducted to determine the food and feeding habits of blue peafowl in three study sites, namely, Saraswati plantation wildlife sanctuary (SPWS), Bir Sonti Reserve Forest (BSRF), and Jhrouli Kalan village (JKAL). Point count method (Blondel et al., 1981) was followed during periodic fortnightly visits to all the three selected study sites. The peafowls were observed to feed on flowers, fruits, leaves of 11, 8 and 8 plant species respectively. These were sighted to feed on Brassica compestris (flowers, leaves), Trifolium alexandarium (flowers, leaves), Triticum aestivum (flowers, leaves, fruits), Oryza sativa (flowers, leaves, fruits), Chenopodium album (flowers, leaves, fruits), Parthenium histerophoresus (flowers, leaves), Pisum sativum (flowers, leaves, fruits), Cicer arientum (flowers, leaves, fruits), Pyrus pyrifolia (flowers, fruits), Ficus benghalensis (flowers, fruits), Ficus rumphii (flowers, fruits). They were also observed feeding on insects in all three study sites and on remains of the snake bodies at the BSRF and JKAL study site. The findings revealed that the Indian peafowl, on one hand, functions as a predator of agricultural pests but, on the other hand, is itself a pest on agricultural crops. Keywords: Blue peafowl, Food, Feeding Habits, Herbs, Shrubs, Trees. 1. INTRODUCTION Birds are warm-blooded, bipedal, oviparous vertebrates characterized by bony beak, pneumatic bones, feathers and wings. -

Hybridization & Zoogeographic Patterns in Pheasants
University of Nebraska - Lincoln DigitalCommons@University of Nebraska - Lincoln Paul Johnsgard Collection Papers in the Biological Sciences 1983 Hybridization & Zoogeographic Patterns in Pheasants Paul A. Johnsgard University of Nebraska-Lincoln, [email protected] Follow this and additional works at: https://digitalcommons.unl.edu/johnsgard Part of the Ornithology Commons Johnsgard, Paul A., "Hybridization & Zoogeographic Patterns in Pheasants" (1983). Paul Johnsgard Collection. 17. https://digitalcommons.unl.edu/johnsgard/17 This Article is brought to you for free and open access by the Papers in the Biological Sciences at DigitalCommons@University of Nebraska - Lincoln. It has been accepted for inclusion in Paul Johnsgard Collection by an authorized administrator of DigitalCommons@University of Nebraska - Lincoln. HYBRIDIZATION & ZOOGEOGRAPHIC PATTERNS IN PHEASANTS PAUL A. JOHNSGARD The purpose of this paper is to infonn members of the W.P.A. of an unusual scientific use of the extent and significance of hybridization among pheasants (tribe Phasianini in the proposed classification of Johnsgard~ 1973). This has occasionally occurred naturally, as for example between such locally sympatric species pairs as the kalij (Lophura leucol11elana) and the silver pheasant (L. nycthelnera), but usually occurs "'accidentally" in captive birds, especially in the absence of conspecific mates. Rarely has it been specifically planned for scientific purposes, such as for obtaining genetic, morphological, or biochemical information on hybrid haemoglobins (Brush. 1967), trans ferins (Crozier, 1967), or immunoelectrophoretic comparisons of blood sera (Sato, Ishi and HiraI, 1967). The literature has been summarized by Gray (1958), Delacour (1977), and Rutgers and Norris (1970). Some of these alleged hybrids, especially those not involving other Galliformes, were inadequately doculnented, and in a few cases such as a supposed hybrid between domestic fowl (Gallus gal/us) and the lyrebird (Menura novaehollandiae) can be discounted. -

Europe's Huntable Birds a Review of Status and Conservation Priorities
FACE - EUROPEAN FEDERATIONEurope’s FOR Huntable HUNTING Birds A Review AND CONSERVATIONof Status and Conservation Priorities Europe’s Huntable Birds A Review of Status and Conservation Priorities December 2020 1 European Federation for Hunting and Conservation (FACE) Established in 1977, FACE represents the interests of Europe’s 7 million hunters, as an international non-profit-making non-governmental organisation. Its members are comprised of the national hunters’ associations from 37 European countries including the EU-27. FACE upholds the principle of sustainable use and in this regard its members have a deep interest in the conservation and improvement of the quality of the European environment. See: www.face.eu Reference Sibille S., Griffin, C. and Scallan, D. (2020) Europe’s Huntable Birds: A Review of Status and Conservation Priorities. European Federation for Hunting and Conservation (FACE). https://www.face.eu/ 2 Europe’s Huntable Birds A Review of Status and Conservation Priorities Executive summary Context Non-Annex species show the highest proportion of ‘secure’ status and the lowest of ‘threatened’ status. Taking all wild birds into account, The EU State of Nature report (2020) provides results of the national the situation has deteriorated from the 2008-2012 to the 2013-2018 reporting under the Birds and Habitats directives (2013 to 2018), and a assessments. wider assessment of Europe’s biodiversity. For FACE, the findings are of key importance as they provide a timely health check on the status of In the State of Nature report (2020), ‘agriculture’ is the most frequently huntable birds listed in Annex II of the Birds Directive. -

Alectoris Chukar
PEST RISK ASSESSMENT Chukar partridge Alectoris chukar (Photo: courtesy of Olaf Oliviero Riemer. Image from Wikimedia Commons under a Creative Commons Attribution License, Version 3.) March 2011 This publication should be cited as: Latitude 42 (2011) Pest Risk Assessment: Chukar partridge (Alectoris chukar). Latitude 42 Environmental Consultants Pty Ltd. Hobart, Tasmania. About this Pest Risk Assessment This pest risk assessment is developed in accordance with the Policy and Procedures for the Import, Movement and Keeping of Vertebrate Wildlife in Tasmania (DPIPWE 2011). The policy and procedures set out conditions and restrictions for the importation of controlled animals pursuant to s32 of the Nature Conservation Act 2002. For more information about this Pest Risk Assessment, please contact: Wildlife Management Branch Department of Primary Industries, Parks, Water and Environment Address: GPO Box 44, Hobart, TAS. 7001, Australia. Phone: 1300 386 550 Email: [email protected] Visit: www.dpipwe.tas.gov.au Disclaimer The information provided in this Pest Risk Assessment is provided in good faith. The Crown, its officers, employees and agents do not accept liability however arising, including liability for negligence, for any loss resulting from the use of or reliance upon the information in this Pest Risk Assessment and/or reliance on its availability at any time. Pest Risk Assessment: Chukar partridge Alectoris chukar 2/20 1. Summary The chukar partridge (Alectoris chukar) is native to the mountainous regions of Asia, Western Europe and the Middle East (Robinson 2007, Wikipedia 2009). Its natural range includes Turkey, the Mediterranean islands, Iran and east through Russia and China and south into Pakistan and Nepal (Cowell 2008). -

Banding Cytogenetics of the Barbary Partridge Alectoris Barbara and the Chukar Partridge Alectoris Chukar (Phasianidae): a Large
COMPARATIVE A peer-reviewed open-access journal CompCytogenBanding 12(2): 171–199 cytogenetics (2018) of Alectoris barbara and Alectoris chukar (Phasianidae)... 171 doi: 10.3897/CompCytogen.v12i2.23743 RESEARCH ARTICLE Cytogenetics http://compcytogen.pensoft.net International Journal of Plant & Animal Cytogenetics, Karyosystematics, and Molecular Systematics Banding cytogenetics of the Barbary partridge Alectoris barbara and the Chukar partridge Alectoris chukar (Phasianidae): a large conservation with Domestic fowl Gallus domesticus revealed by high resolution chromosomes Siham Ouchia-Benissad1, Kafia Ladjali-Mohammedi1 1 University of Sciences and Technology Houari Boumediene, Faculty of Biological Sciences, LBCM lab., Team: Genetics of Development. USTHB, PO box 32 El-Alia, Bab-Ezzouar, 16110 Algiers, Algeria Corresponding author: Siham Ouchia-Benissad ([email protected]) Academic editor: S. Galkina | Received 19 January 2018 | Accepted 16 May 2018 | Published 4 June 2018 http://zoobank.org/020C43BA-E325-4B5E-8A17-87358D1B68A5 Citation: Ouchia-Benissad S, Ladjali-Mohammedi K (2018) Banding cytogenetics of the Barbary partridge Alectoris barbara and the Chukar partridge Alectoris chukar (Phasianidae): a large conservation with Domestic fowl Gallus domesticus revealed by high resolution chromosomes. Comparative Cytogenetics 12(2): 171–199. https://doi.org/10.3897/ CompCytogen.v12i2.23743 Abstract The development of avian cytogenetics is significantly behind that of mammals. In fact, since the advent of cytogenetic techniques, fewer than 1500 karyotypes have been established. The Barbary partridge Alec- toris barbara Bonnaterre, 1790 is a bird of economic interest but its genome has not been studied so far. This species is endemic to North Africa and globally declining. The Chukar partridge Alectoris chukar Gray, 1830 is an introduced species which shares the same habitat area as the Barbary partridge and so there could be introgressive hybridisation. -

FULL ACCOUNT FOR: Alectoris Chukar Global Invasive Species Database (GISD) 2021. Species Profile Alectoris Chukar. Available
FULL ACCOUNT FOR: Alectoris chukar Alectoris chukar System: Terrestrial Kingdom Phylum Class Order Family Animalia Chordata Aves Galliformes Phasianidae Common name iwashako (Japanese), chucor (English), coturnice orientale (Italian), perdrix choukar (French), chukar (English), chukarhuhn (German), orebice cukar (Czech), berghöna (Swedish), perdiz chucar (Spanish), vuoripyy (Finnish), aziatische steenpatrijs (Dutch), perdiz- chukar (Portuguese), kuropta cukar (Slovak), chukor (English), Indian chukor (English), chukar partridge (English), chukor partridge (English), rock partridge (English), chukarhøne (Danish), góropatwa azjatycka (Polish), berghæna (Icelandic), berghøne (Norwegian) Synonym Alectoris kakelik Tetrao kakelik Similar species Summary Alectoris chukar has a wide distribution, stretching from the Aegean Sea through to Central and Eastern Asia. There does however seem to be two genetic clades within the species, those from the Mediterranean through to Central Asia and those from Eastern Asia. This is important as individuals used in the introduction into North America and Hawaii were from individuals from Eastern Asia; whereas individuals causing hybrization problems in Europe come from the Mediterranean and Central Asian clade. This hybridization is causing major problems to the genetic purity of the native Alectoris rufa in the Iberian Peninsula, and strict measures in regards to potential hybridization, and the importation and introduction of farm-reared individuals needs to be introduced. view this species on IUCN Red List Notes Alectoris chukar has a wide distribution, stretching from the Aegean Sea through to Central and Eastern Asia (Barbanera et al, 2009b). There does however seem to be two genetic clades within the species, those from the Mediterranean through to Central Asia and those from Eastern Asia (Barbanera et al, 2009b). -

Phylogenetic Relationships of the Phasianidae Reveals Possible Non-Pheasant Taxa
Journal of Heredity 2003:94(6):472–489 Ó 2003 The American Genetic Association DOI: 10.1093/jhered/esg092 Phylogenetic Relationships of the Phasianidae Reveals Possible Non-Pheasant Taxa K. L. BUSH AND C. STROBECK From the Department of Biological Sciences, University of Alberta–Edmonton, Edmonton, Alberta T6G 2E9, Canada. Address correspondence to Krista Bush at the address above, or e-mail: [email protected]. Abstract The phylogenetic relationships of 21 pheasant and 6 non-pheasant species were determined using nucleotide sequences from the mitochondrial cytochrome b gene. Maximum parsimony and maximum likelihood analysis were used to try to resolve the phylogenetic relationships within Phasianidae. Both the degree of resolution and strength of support are improved over previous studies due to the testing of a number of species from multiple pheasant genera, but several major ambiguities persist. Polyplectron bicalcaratum (grey peacock pheasant) is shown not to be a pheasant. Alternatively, it appears ancestral to either the partridges or peafowl. Pucrasia macrolopha macrolopha (koklass) and Gallus gallus (red jungle fowl) both emerge as non-pheasant genera. Monophyly of the pheasant group is challenged if Pucrasia macrolopha macrolopha and Gallus gallus are considered to be pheasants. The placement of Catreus wallichii (cheer) within the pheasants also remains undetermined, as does the cause for the great sequence divergence in Chrysolophus pictus obscurus (black-throated golden). These results suggest that alterations in taxonomic -

Rapid Differentiation of Alectoris Rufa L., 1758 and Alectoris Chukar (Gray, 1830) (Galliformes: Phasianidae) by Melting Curve A
Anales de Biología 36: 121-127, 2014 ARTICLE DOI: http://dx.doi.org/10.6018/analesbio.36.19 Rapid differentiation of Alectoris rufa L., 1758 and Alectoris chukar (Gray, 1830) (Galliformes: Phasianidae) by melting curve analysis of a parathyroid hormone gene SNP María Juliana Rodríguez-García & José Galián Departamento de Zoología y Antropología Física, Facultad de Veterinaria, Campus de Espinardo, Universidad de Murcia, 30100 Murcia, Spain. Resumen Correspondence Diferenciación rápida de Alectoris rufa Linnaeus, 1758 y Alectoris M. J. Rodríguez-García chukar (Gray, 1830) (Galliformes: Phasianidae) mediante curvas E-mail: [email protected] de disociación de un SNP del gen de la hormona paratiroidea Tel. +34 868 888031 Los métodos actuales basados en ADN para identificar los niveles Fax +34 868 884906 de introgresión en especies cercanas implican la manipulación Received: 4 July 2014 posterior a la PCR, aumentando el tiempo y coste en el procesado Accepted: 31 October 2014 de las muestras. El análisis de curvas de disociación es una técni- Published on-line: 20 November 2014 ca que utiliza cebadores especie-específicos. En este trabajo se demuestra que tal análisis resulta muy preciso para la diferencia- ción de individuos puros de Alectoris chukar y A. rufa, utilizando un SNP del gen de la hormona paratiroidea de una manera simple, rá- pida y eficiente. Se analizaron setenta y ocho muestras (A. chukar, n=15; A. rufa, n=63), y no se detectaron híbridos. Este método po- dría ser aplicado en políticas de gestión basadas en el control ge- nético de las perdices reproducidas en granjas y utilizadas en re- población u otras actividades cinegéticas. -
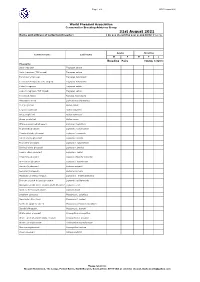
31St August 2021 Name and Address of Collection/Breeder: Do You Closed Ring Your Young Birds? Yes / No
Page 1 of 3 WPA Census 2021 World Pheasant Association Conservation Breeding Advisory Group 31st August 2021 Name and address of collection/breeder: Do you closed ring your young birds? Yes / No Adults Juveniles Common name Latin name M F M F ? Breeding Pairs YOUNG 12 MTH+ Pheasants Satyr tragopan Tragopan satyra Satyr tragopan (TRS ringed) Tragopan satyra Temminck's tragopan Tragopan temminckii Temminck's tragopan (TRT ringed) Tragopan temminckii Cabot's tragopan Tragopan caboti Cabot's tragopan (TRT ringed) Tragopan caboti Koklass pheasant Pucrasia macrolopha Himalayan monal Lophophorus impeyanus Red junglefowl Gallus gallus Ceylon junglefowl Gallus lafayettei Grey junglefowl Gallus sonneratii Green junglefowl Gallus varius White-crested kalij pheasant Lophura l. hamiltoni Nepal Kalij pheasant Lophura l. leucomelana Crawfurd's kalij pheasant Lophura l. crawfurdi Lineated kalij pheasant Lophura l. lineata True silver pheasant Lophura n. nycthemera Berlioz’s silver pheasant Lophura n. berliozi Lewis’s silver pheasant Lophura n. lewisi Edwards's pheasant Lophura edwardsi edwardsi Vietnamese pheasant Lophura e. hatinhensis Swinhoe's pheasant Lophura swinhoii Salvadori's pheasant Lophura inornata Malaysian crestless fireback Lophura e. erythrophthalma Bornean crested fireback pheasant Lophura i. ignita/nobilis Malaysian crestless fireback/Vieillot's Pheasant Lophura i. rufa Siamese fireback pheasant Lophura diardi Southern Cavcasus Phasianus C. colchicus Manchurian Ring Neck Phasianus C. pallasi Northern Japanese Green Phasianus versicolor -

Patterns of Success in Game Bird Introductions in the United States
February 18, 2016 1 Patterns of Success in Game Bird Introductions in the United States 2 3 4 by 5 6 7 8 1Michael P. Moulton, 2Wendell P. Cropper, Jr., 1Andrew J. Broz, 2Salvador A. 9 Gezan 10 11 1Department of Wildlife Ecology and Conservation; University of Florida; PO 12 Box 110430; Gainesville, FL 32611-0430; <[email protected]> 13 2School of Forest Resources and Conservation; University of Florida; PO Box 14 110410; Gainesville, FL 32611-0410; <[email protected]> 15 16 "There is no safety in numbers, or in anything else." James Thurber, The Fairly 17 Intelligent Fly, The New Yorker, February 4, 1939. 18 19 20 1 PeerJ PrePrints | https://doi.org/10.7287/peerj.preprints.1765v1 | CC-BY 4.0 Open Access | rec: 20 Feb 2016, publ: 20 Feb 2016 February 18, 2016 21 Abstract 22 Better predictions of the success of species’ introductions require careful 23 evaluation of the relative importance of at least three kinds of factors: species 24 characteristics, characteristics of the site of introduction, and event-level factors 25 such as propagule pressure. Historical records of bird introductions provide a 26 unique method for addressing the relative importance of these factors. We compiled 27 a list of introductions of 17 Phasianid species released in the USA during the Foreign 28 Game Investigation Program (FGIP). These records indicate that releases for some 29 Phasianid species in the USA continued long after establishment. For many of the 13 30 species that always failed, even numerous releases and large numbers of individuals 31 per release were not enough for successful establishment, yet several of these 32 species were successfully introduced elsewhere.