Locus-Specific Epigenetic Remodeling Controls Addiction- and Depression-Related Behaviors
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Epigenetic Regulation of the PTEN–AKT–RAC1 Axis by G9a Is Critical for Tumor Growth in Alveolar Rhabdomyosarcoma Akshay V
Published OnlineFirst March 4, 2019; DOI: 10.1158/0008-5472.CAN-18-2676 Cancer Molecular Cell Biology Research Epigenetic Regulation of the PTEN–AKT–RAC1 Axis by G9a Is Critical for Tumor Growth in Alveolar Rhabdomyosarcoma Akshay V. Bhat1, Monica Palanichamy Kala1, Vinay Kumar Rao1, Luca Pignata2, Huey Jin Lim3, Sudha Suriyamurthy1, Kenneth T.Chang4,Victor K. Lee3, Ernesto Guccione2, and Reshma Taneja1 Abstract Alveolar rhabdomyosarcoma (ARMS) is an aggressive suppressor PTEN was a direct target gene of G9a. G9a pediatric cancer with poor prognosis. As transient and stable repressed PTEN expression in a methyltransferase activity– modifications to chromatin have emerged as critical dependent manner, resulting in increased AKT and RAC1 mechanisms in oncogenic signaling, efforts to target epige- activity. Re-expression of constitutively active RAC1 in G9a- netic modifiers as a therapeutic strategy have accelerated in deficient tumor cells restored oncogenic phenotypes, demon- recent years. To identify chromatin modifiers that sustain strating its critical functions downstream of G9a. Collectively, tumor growth, we performed an epigenetic screen and our study provides evidence for a G9a-dependent epigenetic found that inhibition of lysine methyltransferase G9a sig- program that regulates tumor growth and suggests targeting nificantly affected the viability of ARMS cell lines. Targeting G9a as a therapeutic strategy in ARMS. expression or activity of G9a reduced cellular proliferation and motility in vitro and tumor growth in vivo.Transcrip- Significance: These findings demonstrate that RAC1 is an tome and chromatin immunoprecipitation–sequencing effector of G9a oncogenic functions and highlight the poten- analysis provided mechanistic evidence that the tumor- tial of G9a inhibitors in the treatment of ARMS. -

Overexpression of the Histone Dimethyltransferase G9a in Nucleus Accumbens Shell Increases Cocaine Self- Administration, Stress-Induced Reinstatement, and Anxiety
The Journal of Neuroscience, January 24, 2018 • 38(4):803–813 • 803 Neurobiology of Disease Overexpression of the Histone Dimethyltransferase G9a in Nucleus Accumbens Shell Increases Cocaine Self- Administration, Stress-Induced Reinstatement, and Anxiety X Ethan M. Anderson,1 Erin B. Larson,1 XDaniel Guzman,1 Anne Marie Wissman,1 Rachael L. Neve,2 XEric J. Nestler,3 and X David W. Self1 1Department of Psychiatry, The Seay Center for Basic and Applied Research in Psychiatric Illness, University of Texas Southwestern Medical Center, Dallas, Texas, 75390, 2Viral Gene Transfer Core, Department of Brain and Cognitive Sciences, McGovern Institute for Brain Research, Massachusetts Institute of Technology, Cambridge, Massachusetts 02139, and 3Icahn School of Medicine at Mount Sinai, Department of Neuroscience, New York, New York 10029 Repeated exposure to cocaine induces lasting epigenetic changes in neurons that promote the development and persistence of addiction. One epigenetic alteration involves reductions in levels of the histone dimethyltransferase G9a in nucleus accumbens (NAc) after chronic cocaine administration. This reduction in G9a may enhance cocaine reward because overexpressing G9a in the NAc decreases cocaine- conditioned place preference. Therefore, we hypothesized that HSV-mediated G9a overexpression in the NAc shell (NAcSh) would attenuate cocaine self-administration (SA) and cocaine-seeking behavior. Instead, we found that G9a overexpression, and the resulting increase in histone 3 lysine 9 dimethylation (H3K9me2), increases sensitivity to cocaine reinforcement and enhances motivation for cocaine in self-administering male rats. Moreover, when G9a overexpression is limited to the initial 15 d of cocaine SA training, it produces an enduring postexpression enhancement in cocaine SA and prolonged (over 5 weeks) increases in reinstatement of cocaine seeking induced by foot-shock stress, but in the absence of continued global elevations in H3K9me2. -

A Computational Approach for Defining a Signature of Β-Cell Golgi Stress in Diabetes Mellitus
Page 1 of 781 Diabetes A Computational Approach for Defining a Signature of β-Cell Golgi Stress in Diabetes Mellitus Robert N. Bone1,6,7, Olufunmilola Oyebamiji2, Sayali Talware2, Sharmila Selvaraj2, Preethi Krishnan3,6, Farooq Syed1,6,7, Huanmei Wu2, Carmella Evans-Molina 1,3,4,5,6,7,8* Departments of 1Pediatrics, 3Medicine, 4Anatomy, Cell Biology & Physiology, 5Biochemistry & Molecular Biology, the 6Center for Diabetes & Metabolic Diseases, and the 7Herman B. Wells Center for Pediatric Research, Indiana University School of Medicine, Indianapolis, IN 46202; 2Department of BioHealth Informatics, Indiana University-Purdue University Indianapolis, Indianapolis, IN, 46202; 8Roudebush VA Medical Center, Indianapolis, IN 46202. *Corresponding Author(s): Carmella Evans-Molina, MD, PhD ([email protected]) Indiana University School of Medicine, 635 Barnhill Drive, MS 2031A, Indianapolis, IN 46202, Telephone: (317) 274-4145, Fax (317) 274-4107 Running Title: Golgi Stress Response in Diabetes Word Count: 4358 Number of Figures: 6 Keywords: Golgi apparatus stress, Islets, β cell, Type 1 diabetes, Type 2 diabetes 1 Diabetes Publish Ahead of Print, published online August 20, 2020 Diabetes Page 2 of 781 ABSTRACT The Golgi apparatus (GA) is an important site of insulin processing and granule maturation, but whether GA organelle dysfunction and GA stress are present in the diabetic β-cell has not been tested. We utilized an informatics-based approach to develop a transcriptional signature of β-cell GA stress using existing RNA sequencing and microarray datasets generated using human islets from donors with diabetes and islets where type 1(T1D) and type 2 diabetes (T2D) had been modeled ex vivo. To narrow our results to GA-specific genes, we applied a filter set of 1,030 genes accepted as GA associated. -

Original Article Preprogramming Therapeutic Response of PI3K/Mtor Dual Inhibitor Via the Regulation of EHMT2 and P27 in Pancreatic Cancer
Am J Cancer Res 2018;8(9):1812-1822 www.ajcr.us /ISSN:2156-6976/ajcr0082503 Original Article Preprogramming therapeutic response of PI3K/mTOR dual inhibitor via the regulation of EHMT2 and p27 in pancreatic cancer Yu-Feng Tian1,2, Hui-Ching Wang3,4, Chi-Wen Luo5, Wen-Chun Hung6, Yu-Han Lin3, Tzu-Yi Chen3, Chien-Feng Li7,8,9, Chen-Yi Lin10, Mei-Ren Pan3 1Division of Colorectal Surgery, Department of Surgery, Chi-Mei Medical Center, Tainan, Taiwan; 2Department of Health and Nutrition, Chia Nan University of Pharmacy and Science, Tainan, Taiwan; 3Graduate Institute of Clinical Medicine, College of Medicine, Kaohsiung Medical University, Kaohsiung, Taiwan; 4Division of Hematology and Oncology, Department of Internal Medicine, Kaohsiung Medical University Hospital, Kaohsiung Medical University, Kaohsiung, Taiwan; 5Division of Cardiology, Chang Gung Memorial Hospital-Kaohsiung Medical Center, Kaohsiung, Taiwan; 6National Institute of Cancer Research, National Health Research Institutes, Tainan 704, Taiwan; 7De- partment of Pathology, Chi Mei Medical Center, Tainan, Taiwan; 8National Institute of Cancer Research, National Health Research Institutes, Tainan, Taiwan; 9Department of Biotechnology, Southern Taiwan University of Science and Technology, Tainan, Taiwan; 10Division of Gastroenterology and Hepatology, Department of Internal Medicine, Chi-Mei Medical Center, Tainan, Taiwan Received July 12, 2018; Accepted August 14, 2018; Epub September 1, 2018; Published September 15, 2018 Abstract: Pancreatic ductal adenocarcinoma (PDAC) is an aggressive disease, which is characterized by its high in- vasiveness, rapid progression, and profound resistance to therapy. Gemcitabine is the first-line treatment option for pancreatic cancer patients, but the overall survival is quite low. Therefore, it is an urgent issue to identify new mol- ecules for improved therapies, with better efficacy and less toxicity. -

Development and Validation of a Protein-Based Risk Score for Cardiovascular Outcomes Among Patients with Stable Coronary Heart Disease
Supplementary Online Content Ganz P, Heidecker B, Hveem K, et al. Development and validation of a protein-based risk score for cardiovascular outcomes among patients with stable coronary heart disease. JAMA. doi: 10.1001/jama.2016.5951 eTable 1. List of 1130 Proteins Measured by Somalogic’s Modified Aptamer-Based Proteomic Assay eTable 2. Coefficients for Weibull Recalibration Model Applied to 9-Protein Model eFigure 1. Median Protein Levels in Derivation and Validation Cohort eTable 3. Coefficients for the Recalibration Model Applied to Refit Framingham eFigure 2. Calibration Plots for the Refit Framingham Model eTable 4. List of 200 Proteins Associated With the Risk of MI, Stroke, Heart Failure, and Death eFigure 3. Hazard Ratios of Lasso Selected Proteins for Primary End Point of MI, Stroke, Heart Failure, and Death eFigure 4. 9-Protein Prognostic Model Hazard Ratios Adjusted for Framingham Variables eFigure 5. 9-Protein Risk Scores by Event Type This supplementary material has been provided by the authors to give readers additional information about their work. Downloaded From: https://jamanetwork.com/ on 10/02/2021 Supplemental Material Table of Contents 1 Study Design and Data Processing ......................................................................................................... 3 2 Table of 1130 Proteins Measured .......................................................................................................... 4 3 Variable Selection and Statistical Modeling ........................................................................................ -

Evidence for Non-Genetic Factors in C57BL/6 Mice Jennifer T
Virginia Commonwealth University VCU Scholars Compass Pharmacology and Toxicology Publications Dept. of Pharmacology and Toxicology 2011 Genomic Analysis of Individual Differences in Ethanol Drinking: Evidence for Non-Genetic Factors in C57BL/6 Mice Jennifer T. Wolstenholme Virginia Commonwealth University, [email protected] Jon A. Warner Virginia Commonwealth University Maria I. Capparuccini Virginia Commonwealth University, [email protected] See next page for additional authors Follow this and additional works at: http://scholarscompass.vcu.edu/phtx_pubs Part of the Medical Pharmacology Commons © 2011 Wolstenholme et al. This is an open-access article distributed under the terms of the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original author and source are credited. Downloaded from http://scholarscompass.vcu.edu/phtx_pubs/9 This Article is brought to you for free and open access by the Dept. of Pharmacology and Toxicology at VCU Scholars Compass. It has been accepted for inclusion in Pharmacology and Toxicology Publications by an authorized administrator of VCU Scholars Compass. For more information, please contact [email protected]. Authors Jennifer T. Wolstenholme, Jon A. Warner, Maria I. Capparuccini, Kellie J. Archer, Keith L. Shelton, and Michael F. Miles This article is available at VCU Scholars Compass: http://scholarscompass.vcu.edu/phtx_pubs/9 Genomic Analysis of Individual Differences in Ethanol Drinking: Evidence for Non-Genetic Factors -

Longitudinal Genome-Wide Methylation Study of Roux-En-Y Gastric Bypass Patients Reveals Novel Cpg Sites Associated with Essential Hypertension Adrian E
Boström et al. BMC Medical Genomics (2016) 9:20 DOI 10.1186/s12920-016-0180-y RESEARCH ARTICLE Open Access Longitudinal genome-wide methylation study of Roux-en-Y gastric bypass patients reveals novel CpG sites associated with essential hypertension Adrian E. Boström1*, Jessica Mwinyi1, Sarah Voisin1, Wenting Wu2, Bernd Schultes3, Kang Zhang2 and Helgi B. Schiöth1 Abstract Background: Essential hypertension is a significant risk factor for cardiovascular diseases. Emerging research suggests a role of DNA methylation in blood pressure physiology. We aimed to investigate epigenetic associations of promoter related CpG sites to essential hypertension in a genome-wide methylation approach. Methods: The genome-wide methylation pattern in whole blood was measured in 11 obese patients before and six months after Roux-en-Y gastric bypass surgery using the Illumina 450 k beadchip. CpG sites located within 1500 bp of the transcriptional start site of adjacent genes were included in our study, resulting in 124 199 probes investigated in the subsequent analysis. Percent changes in methylation states and SBP measured before and six months after surgery were calculated. These parameters were correlated to each other using the Spearman’s rank correlation method (Edgeworth series approximation). To further investigate the detected relationship between candidate CpG sites and systolic blood pressure levels, binary logistic regression analyses were performed in a larger and independent cohort of 539 individuals aged 19–101 years to elucidate a relationship between EH and the methylation state in candidate CpG sites. Results: We identified 24 promoter associated CpG sites that correlated with change in SBP after RYGB surgery (p <10−16). -

A Validation of Illumina EPIC Array System with Bisulfite-Based Amplicon Sequencing
bioRxiv preprint doi: https://doi.org/10.1101/2020.05.25.115428; this version posted May 28, 2020. The copyright holder for this preprint (which was not certified by peer review) is the author/funder, who has granted bioRxiv a license to display the preprint in perpetuity. It is made available under aCC-BY-NC-ND 4.0 International license. A validation of Illumina EPIC array system with bisulfite-based amplicon sequencing Alexandra Noble1, John Pearson2, Joseph Boden3, John Horwood3, Neil Gemmell4, Martin Kennedy2, Amy Osborne1 1Biological Sciences, University of Canterbury, Christchurch, New Zealand 2Department of Pathology and Biomedical Sciences, University of Otago, Christchurch, New Zealand 3Department of Psychological Medicine, University of Otago, Christchurch, New Zealand 4Department of Anatomy, University of Otago, Dunedin, New Zealand bioRxiv preprint doi: https://doi.org/10.1101/2020.05.25.115428; this version posted May 28, 2020. The copyright holder for this preprint (which was not certified by peer review) is the author/funder, who has granted bioRxiv a license to display the preprint in perpetuity. It is made available under aCC-BY-NC-ND 4.0 International license. Abstract The Illumina Infinium® MethylationEPIC BeadChip system (hereafter EPIC array) is considered to be the current gold standard detection method for assessing DNA methylation at the genome-wide level. EPIC arrays are used for hypothesis generation or pilot studies, the natural conclusion is to validate methylation candidates and expand these in a larger cohort, in a targeted manner. As such, an accurate smaller-scale, targeted technique, that generates data at the individual CpG level that is equivalent to the EPIC array, is needed. -

EHMT2 Directs DNA Methylation for Efficient Gene Silencing in Mouse Embryos
Downloaded from genome.cshlp.org on September 30, 2021 - Published by Cold Spring Harbor Laboratory Press Research EHMT2 directs DNA methylation for efficient gene silencing in mouse embryos Ghislain Auclair,1 Julie Borgel,2,3,4 Lionel A. Sanz,2,3,5 Judith Vallet,1 Sylvain Guibert,1 Michael Dumas,1 Patricia Cavelier,2 Michael Girardot,2 Thierry Forné,2 Robert Feil,2 and Michael Weber1 1CNRS, University of Strasbourg, UMR7242 Biotechnology and Cell Signaling, 67412 Illkirch, France; 2Institute of Molecular Genetics, CNRS UMR5535, University of Montpellier, 34293 Montpellier, France The extent to which histone modifying enzymes contribute to DNA methylation in mammals remains unclear. Previous studies suggested a link between the lysine methyltransferase EHMT2 (also known as G9A and KMT1C) and DNA methyl- ation in the mouse. Here, we used a model of knockout mice to explore the role of EHMT2 in DNA methylation during mouse embryogenesis. The Ehmt2 gene is expressed in epiblast cells but is dispensable for global DNA methylation in embryogenesis. In contrast, EHMT2 regulates DNA methylation at specific sequences that include CpG-rich promoters of germline-specific genes. These loci are bound by EHMT2 in embryonic cells, are marked by H3K9 dimethylation, and have strongly reduced DNA methylation in Ehmt2−/− embryos. EHMT2 also plays a role in the maintenance of germ- line-derived DNA methylation at one imprinted locus, the Slc38a4 gene. Finally, we show that DNA methylation is instru- mental for EHMT2-mediated gene silencing in embryogenesis. Our findings identify EHMT2 as a critical factor that facilitates repressive DNA methylation at specific genomic loci during mammalian development. -

B P50 and Represses Gene Expression* S
Supplemental Material can be found at: http://www.jbc.org/content/suppl/2012/07/16/M112.365601.DC1.html THE JOURNAL OF BIOLOGICAL CHEMISTRY VOL. 287, NO. 37, pp. 31207–31217, September 7, 2012 © 2012 by The American Society for Biochemistry and Molecular Biology, Inc. Published in the U.S.A. EHMT1 Protein Binds to Nuclear Factor-B p50 and Represses Gene Expression*□S Received for publication, March 22, 2012, and in revised form, July 14, 2012 Published, JBC Papers in Press, July 16, 2012, DOI 10.1074/jbc.M112.365601 Chee-Kwee Ea‡, ShengLi Hao§, Kok Siong Yeo‡, and David Baltimore§1 From the ‡Institute of Biological Sciences, Faculty of Science, University of Malaya, 50603 Kuala Lumpur, Malaysia and the §Division of Biology, California Institute of Technology, Pasadena, California 91125 Background: Methylation of histone H3 lysine 9 (H3K9) is commonly correlated with gene repression. Results: p50 recruits EHMT1 to repress gene expression. Conclusion: EHMT1 is a negative regulator of NF-B and type I interferon induction pathways. Downloaded from Significance: p50-dependent changes in the chromatin structure importantly contribute to the repression of the expression genes in resting cells. Transcriptional homeostasis relies on the balance between most studied H3K9-HMTs are SUV39H1 and SUV39H2, which positive and negative regulation of gene transcription. Methyla- promote the formation of pericentric heterochromatin (6, 7). www.jbc.org tion of histone H3 lysine 9 (H3K9) is commonly correlated with Methylation of H3K9 by SUV39H1 creates a docking site for gene repression. Here, we report that a euchromatic H3K9 subsequent binding of the heterochromatin protein HP1, pro- methyltransferase, EHMT1, functions as a negative regulator in viding compelling evidence linking H3K9 methylation to het- both the NF- B- and type I interferon-mediated gene induction erochromatin formation and transcriptional repression (8–10). -

Transcriptomic Changes in Mouse Bone Marrow-Derived Macrophages Exposed to Neuropeptide FF
G C A T T A C G G C A T genes Article Transcriptomic Changes in Mouse Bone Marrow-Derived Macrophages Exposed to Neuropeptide FF Yulong Sun 1,2,* , Yuanyuan Kuang 1,2 and Zhuo Zuo 1,2 1 School of Life Sciences, Northwestern Polytechnical University, Xi’an 710072, China; [email protected] (Y.K.); [email protected] (Z.Z.) 2 Key Laboratory for Space Biosciences & Biotechnology, Institute of Special Environmental Biophysics, School of Life Sciences, Northwestern Polytechnical University, Xi’an 710072, China * Correspondence: [email protected]; Tel.: +86-29-8846-0332 Abstract: Neuropeptide FF (NPFF) is a neuropeptide that regulates various biological activities. Currently, the regulation of NPFF on the immune system is an emerging field. However, the influence of NPFF on the transcriptome of primary macrophages has not been fully elucidated. In this study, the effect of NPFF on the transcriptome of mouse bone marrow-derived macrophages (BMDMs) was explored by RNA sequencing, bioinformatics, and molecular simulation. BMDMs were treated with 1 nM NPFF for 18 h, followed by RNA sequencing. Differentially expressed genes (DEGs) were obtained, followed by GO, KEGG, and PPI analysis. A total of eight qPCR-validated DEGs were selected as hub genes. Subsequently, the three-dimensional (3-D) structures of the eight hub proteins were constructed by Modeller and Rosetta. Next, the molecular dynamics (MD)-optimized 3-D structure of hub protein was acquired with Gromacs. Finally, the binding modes between NPFF and hub proteins were studied by Rosetta. A total of 2655 DEGs were obtained (up-regulated 1442 vs. -
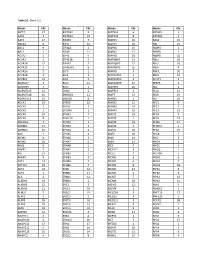
Table S1. Gene List Genes Chr Genes Chr Genes Chr Genes Chr SEPT9
Table S1. Gene List Genes Chr Genes Chr Genes Chr Genes Chr SEPT9 17 EIF2AK3 2 MAPK14 6 RASSF5 1 AAK1 2 EIF2AK4 15 MAPK15 8 RAVER2 1 AATK 17 EIF2B5 3 MAPK3 16 RAX2 19 ABCA1 9 EIF3J 15 MAPK4 18 RB1 13 ABL1 9 EIF4A2 3 MAPK6 15 RBBP4 1 ABL2 1 EIF4B 12 MAPK7 17 RBBP5 1 ACTR2 2 EIF4E 4 MAPK8 10 RBBP8 18 ACVR1 2 EIF4E1B 5 MAPK8IP1 11 RBL1 20 ACVR1B 12 EIF4E2 2 MAPK8IP2 22 RBL2 16 ACVR1C 2 EIF4EBP1 8 MAPK8IP3 16 RBPJ 4 ACVR2A 2 ELF3 1 MAPK9 5 RBPJL 20 ACVR2B 3 ELF4 X MAPKAPK2 1 RBX1 22 ACVRL1 12 ELK1 X MAPKAPK3 3 RCHY1 4 ADAM17 2 ELK3 12 MAPKAPK5 12 REEP5 5 ADAM29 4 ELK4 1 MAPRE1 20 REL 2 ADAMTS15 11 EML4 2 MAPRE3 2 RELA 11 ADAMTS18 16 ENDOD1 11 MAPT 17 RELB 19 ADAMTSL3 15 ENDOG 9 MARK1 1 RET 10 ADCK1 14 EP300 22 MARK2 11 RFC1 4 ADCK2 7 EPAS1 2 MARK3 14 RFC2 7 ADCK3 1 EPCAM 2 MARK4 19 RFC3 13 ADCK4 19 EPHA1 7 MAST1 19 RFC4 3 ADCK5 8 EPHA10 1 MAST2 1 RFC5 12 ADORA1 1 EPHA2 1 MAST3 19 RFNG 17 ADRBK1 11 EPHA3 3 MAST4 5 RFWD2 1 ADRBK2 22 EPHA4 2 MASTL 10 RFX2 19 AGK 7 EPHA5 4 MATK 19 RHEB 7 AIFM1 X EPHA6 3 MAX 14 RHO 3 AIFM2 10 EPHA7 6 MBIP 14 RHOA 3 AIM1 6 EPHA8 1 MCC 5 RHOC 1 AIMP2 7 EPHB1 3 MCF2L2 3 RHOQ 2 AIP 11 EPHB2 1 MCL1 1 RICTOR 5 AKAP4 X EPHB3 3 MCM2 3 RIOK1 6 AKT1 14 EPHB4 7 MCM3 6 RIOK2 5 AKT1S1 19 EPHB6 7 MCM4 8 RIOK3 18 AKT2 19 EPS8 12 MCM5 22 RIPK1 6 AKT3 1 ERBB2 17 MCM6 2 RIPK2 8 ALK 2 ERBB3 12 MCM7 7 RIPK3 14 ALKBH1 14 ERBB4 2 MCM8 20 RIPK4 21 ALKBH2 12 ERC2 3 MDM2 12 RMI1 9 ALKBH3 11 ERCC1 19 MDM4 1 RNASEL 1 ALMS1 2 ERCC2 19 MECOM 3 RNF213 17 ALPK1 4 ERCC3 2 MED12 X RNF220 1 ALPK2 18 ERCC4 16 MED12L 3 ROCK1 18 ALPK3 15 ERCC5 13