Identification of a Novel Mutation Disrupting the DNA Binding Activity
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Human Stem Cells from Single Blastomeres Reveal Pathways of Embryonic Or Trophoblast Fate Specification Tamara Zdravkovic1,2,3,4,5,‡, Kristopher L
© 2015. Published by The Company of Biologists Ltd | Development (2015) 142, 4010-4025 doi:10.1242/dev.122846 STEM CELLS AND REGENERATION RESEARCH ARTICLE Human stem cells from single blastomeres reveal pathways of embryonic or trophoblast fate specification Tamara Zdravkovic1,2,3,4,5,‡, Kristopher L. Nazor6,‡, Nicholas Larocque1,2,3,4,5, Matthew Gormley1,2,3,4,5, Matthew Donne1,2,3,7, Nathan Hunkapillar1,2,3,4,5, Gnanaratnam Giritharan8, Harold S. Bernstein4,9, Grace Wei4,10, Matthias Hebrok10, Xianmin Zeng11, Olga Genbacev1,2,3,4,5, Aras Mattis4,12, Michael T. McMaster4,5,13, Ana Krtolica8,*, Diana Valbuena14, Carlos Simón14, Louise C. Laurent6,15, Jeanne F. Loring6 and Susan J. Fisher1,2,3,4,5,7,§ ABSTRACT INTRODUCTION For many reasons, relatively little is known about human Mechanisms of initial cell fate decisions differ among species. To gain preimplantation development. The small number of cells makes insights into lineage allocation in humans, we derived ten human embryos of any species difficult to study. In humans, the technical embryonic stem cell lines (designated UCSFB1-10) from single difficulties are compounded by other challenges. Genetic variation blastomeres of four 8-cell embryos and one 12-cell embryo from a among individuals could contribute to developmental differences, a single couple. Compared with numerous conventional lines from well-appreciated phenomenon in the mouse (Dackor et al., 2009), blastocysts, they had unique gene expression and DNA methylation which is difficult to assess in humans owing to the limited patterns that were, in part, indicative of trophoblast competence. At a availability of embryos that are donated for research. -

Primepcr™Assay Validation Report
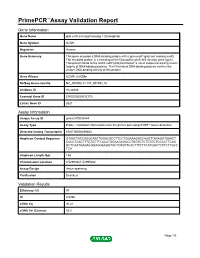
PrimePCR™Assay Validation Report Gene Information Gene Name glial cells missing homolog 2 (Drosophila) Gene Symbol GCM2 Organism Human Gene Summary This gene is a homolog of the Drosophila glial cells missing gene which is thought to act as a binary switch between neuronal and glial cell determination. The protein encoded by this gene contains a conserved N-terminal GCM motif that has DNA-binding activity. The protein is a transcription factor that acts as a master regulator of parathyroid development. It has been suggested that this transcription factor might mediate the effect of calcium on parathyroid hormone expression and secretion in parathyroid cells. Mutations in this gene are associated with hypoparathyroidism. Gene Aliases GCMB, hGCMb RefSeq Accession No. NC_000006.11, NG_008970.1, NT_007592.15 UniGene ID Hs.227098 Ensembl Gene ID ENSG00000124827 Entrez Gene ID 9247 Assay Information Unique Assay ID qHsaCIP0029896 Assay Type Probe - Validation information is for the primer pair using SYBR® Green detection Detected Coding Transcript(s) ENST00000379491 Amplicon Context Sequence ACATAGCCGTCAGGCCACTCTCGGAATTGGTCAAAGAGGGCCAGCTCCTGAGG CATCTGCGGATCGTTGATGTCCCAGCTGAGCTGCATCCCGTA Amplicon Length (bp) 65 Chromosome Location 6:10877579-10881984 Assay Design Intron-spanning Purification Desalted Validation Results Efficiency (%) 101 R2 0.9999 cDNA Cq Target not expressed in universal RNA cDNA Tm (Celsius) Target not expressed in universal RNA Page 1/5 PrimePCR™Assay Validation Report gDNA Cq Target not expressed in universal RNA Specificity -

Presence and Significance of a R110W Mutation in the DNA
European Journal of Endocrinology (2010) 162 407–421 ISSN 0804-4643 CLINICAL STUDY Presence and significance of a R110W mutation in the DNA-binding domain of GCM2 gene in patients with isolated hypoparathyroidism and their family members Neeraj Tomar1, Hema Bora2, Ratnakar Singh3, Nandita Gupta1, Punit Kaur4, Shyam Singh Chauhan3, Yagya Dutta Sharma2 and Ravinder Goswami1 Departments of 1Endocrinology and Metabolism, 2Biotechnology, 3Biochemistry and 4Biophysics, All India Institute of Medical Sciences, New Delhi 110029, India (Correspondence should be addressed to R Goswami; Email: [email protected]) Abstract Objective: Glial cells missing 2 (GCM2) gene encodes a parathyroid-specific transcription factor. We assessed GCM2 gene sequence in patients with isolated hypoparathyroidism (IH). Design: Case–control study. Methods: Complete DNA sequencing of the GCM2 gene including its exons, promoter, and 50 and 30 UTRs was performed in 24/101 patients with IH. PCR–restriction fragment length polymorphism was used to detect a novel R110W mutation in all 101 IH patients and 655 healthy controls. Significance of the mutation was assessed by electrophoretic mobility shift assay (EMSA) and nuclear localization on transfection. Results: A heterozygous R110W mutation was present in DNA-binding domain in 11/101 patients (10.9%) and absent in 655 controls (P!10K7). Four of 13 nonaffected first-degree relatives for five of these index cases had R110W mutation. Four heterozygous single nucleotide polymorphisms were found in the 50 region. One of the 11 patients with R110W also had T370M change in compound heterozygous form. Mutant R110W and T370M GCM2 proteins showed decreased binding with GCM recognition elements on EMSA indicating loss of function. -
![View Software Version 1.6.6 [51]](https://docslib.b-cdn.net/cover/1758/view-software-version-1-6-6-51-591758.webp)
View Software Version 1.6.6 [51]
BMC Genomics BioMed Central Research article Open Access Phylogenetic and comparative gene expression analysis of barley (Hordeum vulgare) WRKY transcription factor family reveals putatively retained functions between monocots and dicots Elke Mangelsen1, Joachim Kilian2, Kenneth W Berendzen2, Üner H Kolukisaoglu3, Klaus Harter2, Christer Jansson1,4 and Dierk Wanke*2 Address: 1Department of Plant Biology and Forest Genetics, The Swedish University of Agricultural Sciences (SLU), P.O. Box 7080, SE-750 07 Uppsala, Sweden, 2Center of Plant Molecular Biology (ZMBP), University of Tuebingen, Auf der Morgenstelle 1, D-72076 Tübingen, Germany, 3CELISCA (Center for Life Science Automation), Friedrich-Barnewitz-Strasse 8, D-18119 Rostock, Germany and 4Lawrence Berkeley Laboratory, Earth Science Division, 1 Cyclotron Rd., Berkeley, CA 94720, USA Email: Elke Mangelsen - [email protected]; Joachim Kilian - [email protected]; Kenneth W Berendzen - [email protected]; Üner H Kolukisaoglu - [email protected]; Klaus Harter - [email protected]; Christer Jansson - [email protected]; Dierk Wanke* - [email protected] tuebingen.de * Corresponding author Published: 28 April 2008 Received: 20 October 2007 Accepted: 28 April 2008 BMC Genomics 2008, 9:194 doi:10.1186/1471-2164-9-194 This article is available from: http://www.biomedcentral.com/1471-2164/9/194 © 2008 Mangelsen et al; licensee BioMed Central Ltd. This is an Open Access article distributed under the terms of the Creative Commons Attribution License (http://creativecommons.org/licenses/by/2.0), which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited. Abstract Background: WRKY proteins belong to the WRKY-GCM1 superfamily of zinc finger transcription factors that have been subject to a large plant-specific diversification. -

Supplementary Materials
Supplementary materials Supplementary Table S1: MGNC compound library Ingredien Molecule Caco- Mol ID MW AlogP OB (%) BBB DL FASA- HL t Name Name 2 shengdi MOL012254 campesterol 400.8 7.63 37.58 1.34 0.98 0.7 0.21 20.2 shengdi MOL000519 coniferin 314.4 3.16 31.11 0.42 -0.2 0.3 0.27 74.6 beta- shengdi MOL000359 414.8 8.08 36.91 1.32 0.99 0.8 0.23 20.2 sitosterol pachymic shengdi MOL000289 528.9 6.54 33.63 0.1 -0.6 0.8 0 9.27 acid Poricoic acid shengdi MOL000291 484.7 5.64 30.52 -0.08 -0.9 0.8 0 8.67 B Chrysanthem shengdi MOL004492 585 8.24 38.72 0.51 -1 0.6 0.3 17.5 axanthin 20- shengdi MOL011455 Hexadecano 418.6 1.91 32.7 -0.24 -0.4 0.7 0.29 104 ylingenol huanglian MOL001454 berberine 336.4 3.45 36.86 1.24 0.57 0.8 0.19 6.57 huanglian MOL013352 Obacunone 454.6 2.68 43.29 0.01 -0.4 0.8 0.31 -13 huanglian MOL002894 berberrubine 322.4 3.2 35.74 1.07 0.17 0.7 0.24 6.46 huanglian MOL002897 epiberberine 336.4 3.45 43.09 1.17 0.4 0.8 0.19 6.1 huanglian MOL002903 (R)-Canadine 339.4 3.4 55.37 1.04 0.57 0.8 0.2 6.41 huanglian MOL002904 Berlambine 351.4 2.49 36.68 0.97 0.17 0.8 0.28 7.33 Corchorosid huanglian MOL002907 404.6 1.34 105 -0.91 -1.3 0.8 0.29 6.68 e A_qt Magnogrand huanglian MOL000622 266.4 1.18 63.71 0.02 -0.2 0.2 0.3 3.17 iolide huanglian MOL000762 Palmidin A 510.5 4.52 35.36 -0.38 -1.5 0.7 0.39 33.2 huanglian MOL000785 palmatine 352.4 3.65 64.6 1.33 0.37 0.7 0.13 2.25 huanglian MOL000098 quercetin 302.3 1.5 46.43 0.05 -0.8 0.3 0.38 14.4 huanglian MOL001458 coptisine 320.3 3.25 30.67 1.21 0.32 0.9 0.26 9.33 huanglian MOL002668 Worenine -

Aberrant Gcm1 Expression Mediates Wnt/Β-Catenin Pathway Activation in Folate Deficiency Involved in Neural Tube Defects
Li et al. Cell Death and Disease (2021) 12:234 https://doi.org/10.1038/s41419-020-03313-z Cell Death & Disease ARTICLE Open Access Aberrant Gcm1 expression mediates Wnt/ β-catenin pathway activation in folate deficiency involved in neural tube defects Jianting Li1,QiuXie2,JunGao 3, Fang Wang4,YihuaBao4,LihuaWu4,LihongYang5, Zhizhen Liu1,RuiGuo1, Ajab Khan1, Dan Liu1,CaihuaLi6, Jianxin Wu4 and Jun Xie 1 Abstract Wnt signaling plays a major role in early neural development. An aberrant activation in Wnt/β-catenin pathway causes defective anteroposterior patterning, which results in neural tube closure defects (NTDs). Changes in folate metabolism may participate in early embryo fate determination. We have identified that folate deficiency activated Wnt/β-catenin pathway by upregulating a chorion-specific transcription factor Gcm1. Specifically, folate deficiency promoted formation of the Gcm1/β-catenin/T-cell factor (TCF4) complex formation to regulate the Wnt targeted gene transactivation through Wnt-responsive elements. Moreover, the transcription factor Nanog upregulated Gcm1 transcription in mESCs under folate deficiency. Lastly, in NTDs mouse models and low-folate NTDs human brain samples, Gcm1 and Wnt/β-catenin targeted genes related to neural tube closure are specifically overexpressed. These results indicated that low-folate level promoted Wnt/β-catenin signaling via activating Gcm1, and thus leaded into aberrant vertebrate neural development. 1234567890():,; 1234567890():,; 1234567890():,; 1234567890():,; Introduction embryonic neural stem cells inhibit the proliferation of Neural tube defects (NTDs) are severe birth defects neural progenitor cells and promote their differentiation6. thought to be associated with genetic and environmental Aberrant canonical Wnt/β-catenin pathway signaling factors1, resulted from the failure of neural tube closure leads to defective anteroposterior patterning7. -

GCM1 Antibody - N-Terminal Region Rabbit Polyclonal Antibody Catalog # AI16248
10320 Camino Santa Fe, Suite G San Diego, CA 92121 Tel: 858.875.1900 Fax: 858.622.0609 GCM1 antibody - N-terminal region Rabbit Polyclonal Antibody Catalog # AI16248 Specification GCM1 antibody - N-terminal region - Product Information Application IHC, WB Primary Accession Q9NP62 Other Accession NM_003643, NP_003634 Reactivity Human, Mouse, Rat, Dog Predicted Human, Mouse, Rat, Dog Host Rabbit Clonality Polyclonal Calculated MW 49kDa KDa Human Lung GCM1 antibody - N-terminal region - Additional Information Gene ID 8521 Alias Symbol GCMA, hGCMa Other Names Chorion-specific transcription factor GCMa, hGCMa, GCM motif protein 1, Glial cells missing homolog 1, GCM1, GCMA Format Liquid. Purified antibody supplied in 1x PBS buffer with 0.09% (w/v) sodium azide and Human Spleen 2% sucrose. Reconstitution & Storage Add 50 ul of distilled water. Final anti-GCM1 antibody concentration is 1 mg/ml in PBS buffer with 2% sucrose. For longer periods of storage, store at 20°C. Avoid repeat freeze-thaw cycles. Precautions GCM1 antibody - N-terminal region is for research use only and not for use in diagnostic or therapeutic procedures. WB Suggested Anti-GCM1 Antibody Titration: 0.2-1 μg/ml GCM1 antibody - N-terminal region - Protein ELISA Titer: 1:7812500 Information Positive Control: Human Placenta Page 1/2 10320 Camino Santa Fe, Suite G San Diego, CA 92121 Tel: 858.875.1900 Fax: 858.622.0609 Name GCM1 Synonyms GCMA Function Transcription factor involved in the control of expression of placental growth factor (PGF) and other placenta-specific genes (PubMed:<a href="http://www.uniprot.org/c itations/10542267" target="_blank">10542267</a>, PubMed:<a href="http://www.uniprot.org/ci tations/18160678" Human 293T target="_blank">18160678</a>). -

Detailed Characterization of Human Induced Pluripotent Stem Cells Manufactured for Therapeutic Applications
Stem Cell Rev and Rep DOI 10.1007/s12015-016-9662-8 Detailed Characterization of Human Induced Pluripotent Stem Cells Manufactured for Therapeutic Applications Behnam Ahmadian Baghbaderani 1 & Adhikarla Syama2 & Renuka Sivapatham3 & Ying Pei4 & Odity Mukherjee2 & Thomas Fellner1 & Xianmin Zeng3,4 & Mahendra S. Rao5,6 # The Author(s) 2016. This article is published with open access at Springerlink.com Abstract We have recently described manufacturing of hu- help determine which set of tests will be most useful in mon- man induced pluripotent stem cells (iPSC) master cell banks itoring the cells and establishing criteria for discarding a line. (MCB) generated by a clinically compliant process using cord blood as a starting material (Baghbaderani et al. in Stem Cell Keywords Induced pluripotent stem cells . Embryonic stem Reports, 5(4), 647–659, 2015). In this manuscript, we de- cells . Manufacturing . cGMP . Consent . Markers scribe the detailed characterization of the two iPSC clones generated using this process, including whole genome se- quencing (WGS), microarray, and comparative genomic hy- Introduction bridization (aCGH) single nucleotide polymorphism (SNP) analysis. We compare their profiles with a proposed calibra- Induced pluripotent stem cells (iPSCs) are akin to embryonic tion material and with a reporter subclone and lines made by a stem cells (ESC) [2] in their developmental potential, but dif- similar process from different donors. We believe that iPSCs fer from ESC in the starting cell used and the requirement of a are likely to be used to make multiple clinical products. We set of proteins to induce pluripotency [3]. Although function- further believe that the lines used as input material will be used ally identical, iPSCs may differ from ESC in subtle ways, at different sites and, given their immortal status, will be used including in their epigenetic profile, exposure to the environ- for many years or even decades. -

Primate-Specific Mir-515 Family Members Inhibit Key Genes in Human Trophoblast Differentiation and Are Upregulated in Preeclamps
Primate-specific miR-515 family members inhibit key PNAS PLUS genes in human trophoblast differentiation and are upregulated in preeclampsia Ming Zhanga,1, Sribalasubashini Muralimanoharana, Alison C. Wortmanb, and Carole R. Mendelsona,b,2 aDepartment of Biochemistry, University of Texas Southwestern Medical Center, Dallas, TX 75390-9038; and bDepartment of Obstetrics and Gynecology, University of Texas Southwestern Medical Center, Dallas, TX 75390-9032 Edited by R. Michael Roberts, University of Missouri–Columbia, Columbia, MO, and approved September 29, 2016 (received for review May 20, 2016) Dysregulation of human trophoblast invasion and differentiation can Estrogens formed by placental aromatase may enhance angiogen- result in preeclampsia (PE), a hypertensive disorder of pregnancy with esis, uteroplacental blood flow, and reduce systemic vascular significant morbidity and mortality for mother and offspring. miRNA resistance (10–14). microarray analysis of RNA from human cytotrophoblasts (CytT), In previous studies using transgenic mice and transfected human before and after differentiation to syncytiotrophoblast (SynT) in trophoblasts in primary culture, we identified an ∼300-bp region primary culture, revealed that members of miR-515 family—including upstream of placenta-specific hCYP19A1 exon I.1 that acts as an miR-515-5p, miR-519e-5p, miR-519c-3p, and miR-518f, belonging to enhancesome and is essential for placenta-specific hCYP19A1 the primate- and placenta-specific chromosome 19 miRNA cluster expression. This genomic region contains response elements for es- (C19MC)—were significantly down-regulated upon human SynT dif- trogen receptor-α (ERα) (15), estrogen-related receptor-γ (16), Sp1 ferentiation. The proto-oncogene, c-MYC, which declines during SynT (9), and glial cells missing 1 (GCM1) (17, 18); each of these tran- pri-miR-515-1 differentiation, interacted with E-boxes upstream of scription factors serves a crucial role in placenta-specific hCYP19A1 pri-miR-515-2 and , encoding these mRNAs, to enhance their expres- expression. -

Early Onset Preeclampsia in a Model for Human Placental Trophoblast
Early onset preeclampsia in a model for human placental trophoblast Megan A. Sheridana,b,c,1, Ying Yangb,d,e,1, Ashish Jainf,g, Alex S. Lyonsa,b, Penghua Yangb,h, Sambasiva R. Brahmasanib,i, Aihua Daib, Yuchen Tianb, Mark R. Ellersieckj, Geetu Tutejaf,g, Danny J. Schustk, Laura C. Schulzk, Toshihiko Ezashib,d,1, and R. Michael Robertsa,b,d,2 aDepartment of Biochemistry, University of Missouri, Columbia, MO 65211; bBond Life Sciences Center, University of Missouri, Columbia, MO 65211; cDepartment of Pathology, Centre for Trophoblast Research, University of Cambridge, Cambridge CB2 1QP, United Kingdom; dDivision of Animal Sciences, University of Missouri, Columbia, MO 65211; eDepartment of Molecular Pharmacology and Physiology, University of South Florida, Tampa, FL 33620; fDepartment of Genetics, Development, and Cell Biology, Iowa State University, Ames, IA 50011; gBioinformatics and Computational Biology, Iowa State University, Ames, IA 50011; hDepartment of Obstetrics, Gynecology, and Reproductive Sciences, School of Medicine, University of Maryland, Baltimore, Baltimore, MD 21201; iCentre for Cellular and Molecular Biology (CCMB), Hyderabad 500007, India; jAgriculture Experiment Station, University of Missouri, Columbia, MO 65211; and kDepartment of Obstetrics, Gynecology and Women’s Health, University of Missouri, Columbia, MO 65212 Contributed by R. Michael Roberts, January 8, 2019 (sent for review September 24, 2018; reviewed by Mana M. Parast and Soumen Paul) We describe a model for early onset preeclampsia (EOPE) that uses that provoke endothelial dysfunction and inflammation in the ma- induced pluripotent stem cells (iPSCs) generated from umbilical ternal vessels. In a normal pregnancy, up-regulation of vascular cords of EOPE and control (CTL) pregnancies. -

ADHD) Gene Networks in Children of Both African American and European American Ancestry
G C A T T A C G G C A T genes Article Rare Recurrent Variants in Noncoding Regions Impact Attention-Deficit Hyperactivity Disorder (ADHD) Gene Networks in Children of both African American and European American Ancestry Yichuan Liu 1 , Xiao Chang 1, Hui-Qi Qu 1 , Lifeng Tian 1 , Joseph Glessner 1, Jingchun Qu 1, Dong Li 1, Haijun Qiu 1, Patrick Sleiman 1,2 and Hakon Hakonarson 1,2,3,* 1 Center for Applied Genomics, Children’s Hospital of Philadelphia, Philadelphia, PA 19104, USA; [email protected] (Y.L.); [email protected] (X.C.); [email protected] (H.-Q.Q.); [email protected] (L.T.); [email protected] (J.G.); [email protected] (J.Q.); [email protected] (D.L.); [email protected] (H.Q.); [email protected] (P.S.) 2 Division of Human Genetics, Department of Pediatrics, The Perelman School of Medicine, University of Pennsylvania, Philadelphia, PA 19104, USA 3 Department of Human Genetics, Children’s Hospital of Philadelphia, Philadelphia, PA 19104, USA * Correspondence: [email protected]; Tel.: +1-267-426-0088 Abstract: Attention-deficit hyperactivity disorder (ADHD) is a neurodevelopmental disorder with poorly understood molecular mechanisms that results in significant impairment in children. In this study, we sought to assess the role of rare recurrent variants in non-European populations and outside of coding regions. We generated whole genome sequence (WGS) data on 875 individuals, Citation: Liu, Y.; Chang, X.; Qu, including 205 ADHD cases and 670 non-ADHD controls. The cases included 116 African Americans H.-Q.; Tian, L.; Glessner, J.; Qu, J.; Li, (AA) and 89 European Americans (EA), and the controls included 408 AA and 262 EA. -
Primepcr™Assay Validation Report
PrimePCR™Assay Validation Report Gene Information Gene Name glial cells missing homolog 1 (Drosophila) Gene Symbol GCM1 Organism Human Gene Summary This gene encodes a DNA-binding protein with a gcm-motif (glial cell missing motif). The encoded protein is a homolog of the Drosophila glial cells missing gene (gcm). This protein binds to the GCM-motif (A/G)CCCGCAT a novel sequence among known targets of DNA-binding proteins. The N-terminal DNA-binding domain confers the unique DNA-binding activity of this protein. Gene Aliases GCMA, hGCMa RefSeq Accession No. NC_000006.11, NT_007592.15 UniGene ID Hs.28346 Ensembl Gene ID ENSG00000137270 Entrez Gene ID 8521 Assay Information Unique Assay ID qHsaCIP0030444 Assay Type Probe - Validation information is for the primer pair using SYBR® Green detection Detected Coding Transcript(s) ENST00000259803 Amplicon Context Sequence GTAACTACCAGGCAATTGGACGCCTTCCTGGAAAGACCAAGTTAAAGGTAAACT CCCCTGACTTTGTGTTTCACCTGGAAGAGACCTGGTCTCTGTGCTCCCCTTCAG GCTCAATGAGACGGAGGAAGGTGCTGTGTTCACTTTCTTCATGGCTCTTCTTGCC TCA Amplicon Length (bp) 136 Chromosome Location 6:52993661-52995682 Assay Design Intron-spanning Purification Desalted Validation Results Efficiency (%) 97 R2 0.9996 cDNA Cq 30.21 cDNA Tm (Celsius) 83.5 Page 1/5 PrimePCR™Assay Validation Report gDNA Cq 44.24 Specificity (%) 100 Information to assist with data interpretation is provided at the end of this report. Page 2/5 PrimePCR™Assay Validation Report GCM1, Human Amplification Plot Amplification of cDNA generated from 25 ng of universal reference RNA Melt Peak