Draft Guidance on Nicotine Active Ingredient
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

An Introduction to Fast Dissolving Oral Thin Film Drug Delivery Systems: a Review
Muthadi Radhika Reddy /J. Pharm. Sci. & Res. Vol. 12(7), 2020, 925-940 An Introduction to Fast Dissolving Oral Thin Film Drug Delivery Systems: A Review Muthadi Radhika Reddy1* 1School of pharmacy, Gurunanak Institute of Technical Campus, Hyderabad, Telangana, India and Department of Pharmacy, Gandhi Institute of Technology and Management University, Vizag, Andhra Pradesh, India INTRODUCTION 2. Useful in situations where rapid onset of action Fast dissolving drug delivery systems were first developed required such as in motion sickness, allergic attack, in the late 1970s as an alternative to conventional dosage coughing or asthma forms. These systems consist of solid dosage forms that 3. Has wide range of applications in pharmaceuticals, Rx disintegrate and dissolve quickly in the oral cavity without Prescriptions and OTC medications for treating pain, the need of water [1]. Fast dissolving drug delivery cough/cold, gastro-esophageal reflux disease,erectile systems include orally disintegrating tablets (ODTs) and dysfunction, sleep disorders, dietary supplements, etc oral thin films (OTFs). The Centre for Drug Evaluation [4] and Research (CDER) defines ODTs as,“a solid dosage 4. No water is required for the administration and hence form containing medicinal substances which disintegrates suitable during travelling rapidly, usually within a matter of seconds, when placed 5. Some drugs are absorbed from the mouth, pharynx upon the tongue” [2]. USFDA defines OTFs as, “a thin, and esophagus as the saliva passes down into the flexible, non-friable polymeric film strip containing one or stomach, enhancing bioavailability of drugs more dispersed active pharmaceutical ingredients which is 6. May offer improved bioavailability for poorly water intended to be placed on the tongue for rapid soluble drugs by offering large surface area as it disintegration or dissolution in the saliva prior to disintegrates and dissolves rapidly swallowing for delivery into the gastrointestinal tract” [3]. -

Comprehensive Review on Herbal Toothpaste
Annals of R.S.C.B., ISSN:1583-6258, Vol. 25, Issue 4, 2021, Pages. 9509 - 9518 Received 05 March 2021; Accepted 01 April 2021. Comprehensive Review on Herbal Toothpaste Divya S1, Dr. J Suresh1*, Dr. S. Meenakshi 2 1. Department of Pharmacognosy, JSS College of Pharmacy, JSS Academy of Higher Education & Research, Mysuru-570015 2.Department of Prosthodontics, JSS Dental College, JSS Academy of Higher Education &Research, Mysuru-570015 Corresponding author Dr. J. Suresh Professor Department of Pharmacognosy, JSS College of Pharmacy, JSS Academy of Higher Education & Research, Mysuru-570015 Email: [email protected] Mobile No: 9480197611 ABSTRACT Herbal products for general as well as for oral health care have gained prominence around worldwide. People who aspire towards the use of herbal products often consider these products are relatively safer than products containing synthetic ingredients. Based on increased usage of herbal cosmetics we tried to make a comprehensive review on herbal toothpaste that helps to maintain a proper oral hygiene and free from periodontal disorder, reduce stain, gingivitis, calculus and caries. The present review gives basic information regarding antimicrobial potential of various herbs, formulationexcipients, that can be used in preparation of toothpaste. Key Words: Herbal toothpaste, Anti-microbial screening, Periodontal disorder, Gingivitis, calculus, Dental caries. INTRODUCTION In developing countries, the intensity of infections caused by certain pathogenic micro- organisms that may leads to mortality as well as morbidity in immune-suppressant patients [1]. Multiple abrasives, scent, green lead was used to remove the stain from teeth until mid- 19thcentury. In Medieval period rock salt, fine sand were the key ingredients used by Arabs for tooth cleaning.In the period 1950 AD, Dr. -

Chapter 1 Controlling Drug Delivery
chapter 1 Controlling drug delivery Overview In this chapter we will: & differentiate drug delivery systems according to their physical state & differentiate drug delivery systems according to their route of administration & differentiate drug delivery systems according to their type of drug release & discuss drug transport across epithelial barriers. Introduction KeyPoints & Continued developments in Pharmacotherapy can be defined as the treatment chemistry, molecular biology and prevention of illness and disease by means of and genomics support the drugs of chemical or biological origin. It ranks discovery and developments among the most important methods of medical of new drugs and new drug treatment, together with surgery, physical targets. & treatment, radiation and psychotherapy. There The drug delivery system are many success stories concerning the use of employed can control the pharmacological action of a drugs and vaccines in the treatment, prevention drug, influencing its and in some cases even eradication of diseases pharmacokinetic and (e.g. smallpox, which is currently the only subsequent therapeutic human infectious disease completely profile. eradicated). Although it is almost impossible to estimate the exact extent of the impact of pharmacotherapy on human health, there can be no doubt that pharmacotherapy, together with improved sanitation, better diet and better housing, has improved people’s health, life expectancy and quality of life. Tip Unprecedented developments in genomics Combinatorial chemistry is a way to and molecular biology today offer a plethora of build a variety of structurally related new drug targets. The use of modern chemical drug compounds rapidly and synthetic methods (such as combinatorial systematically. These are assembled chemistry) enables the syntheses of a large from a range of molecular entities number of new drug candidates in shorter times which are put together in different ‘ ’ than ever before. -

WO 2010/044736 Al
(12) INTERNATIONAL APPLICATION PUBLISHED UNDER THE PATENT COOPERATION TREATY (PCT) (19) World Intellectual Property Organization International Bureau (10) International Publication Number (43) International Publication Date 22 April 2010 (22.04.2010) WO 2010/044736 Al (51) International Patent Classification: (74) Agents: Bratt, Henrik et al; Jarnvagsgatan 10 A, S-251 A61K 9/24 (2006.01) A61P 25/34 (2006.01) 10 Helsingborg (SE). (21) International Application Number: (81) Designated States (unless otherwise indicated, for every PCT/SE2009/05 1163 kind of national protection available): AE, AG, AL, AM, AO, AT, AU, AZ, BA, BB, BG, BH, BR, BW, BY, BZ, (22) Date: International Filing CA, CH, CL, CN, CO, CR, CU, CZ, DE, DK, DM, DO, 13 October 2009 (13.10.2009) DZ, EC, EE, EG, ES, FI, GB, GD, GE, GH, GM, GT, (25) Filing Language: English HN, HR, HU, ID, IL, IN, IS, JP, KE, KG, KM, KN, KP, KR, KZ, LA, LC, LK, LR, LS, LT, LU, LY, MA, MD, (26) Publication Language: English ME, MG, MK, MN, MW, MX, MY, MZ, NA, NG, NI, (30) Priority Data: NO, NZ, OM, PE, PG, PH, PL, PT, RO, RS, RU, SC, SD, 0802 189-1 14 October 2008 (14.10.2008) SE SE, SG, SK, SL, SM, ST, SV, SY, TJ, TM, TN, TR, TT, TZ, UA, UG, US, UZ, VC, VN, ZA, ZM, ZW. (71) Applicant (for all designated States except US): McNeil AB [SE/SE]; Box 941, S-25 1 09 Helsingborg (SE). (84) Designated States (unless otherwise indicated, for every kind of regional protection available): ARIPO (BW, GH, (72) Inventors; and GM, KE, LS, MW, MZ, NA, SD, SL, SZ, TZ, UG, ZM, (75) Inventors/Applicants (for US only): LINDELL, Katari- ZW), Eurasian (AM, AZ, BY, KG, KZ, MD, RU, TJ, na [SE/SE]; Skarhult 1325, S-241 93 Eslδv (SE). -
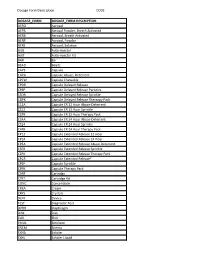
Dosage Form Description CODE
Dosage Form Description CODE DOSAGE_FORM DOSAGE_FORM DESCRIPTION AERO Aerosol AEPB Aerosol Powder, Breath Activated AERB Aerosol, Breath Activated AERP Aerosol, Powder AERS Aerosol, Solution AUIJ Auto-injector AJKT Auto-injector Kit BAR Bar BEAD Beads CAPS Capsule CAPA Capsule Abuse- Deterrent CPCW Capsule Chewable CPDR Capsule Delayed Release CPEP Capsule Delayed Release Particles CSDR Capsule Delayed Release Sprinkle CDPK Capsule Delayed Release Thereapy Pack C12A Capsule ER 12 Hour Abuse-Deterrent CS12 Capsule ER 12 Hour Sprinkle C2PK Capsule ER 12 Hour Therapy Pack C24A Capsule ER 24 Hour Abuse-Deterrent CS24 Capsule ER 24 Hour Sprinkle C4PK Capsule ER 24 Hour Therapy Pack CP12 Capsule Extended Release 12 Hour CP24 Capsule Extended Release 24 Hour CPEA Capsule Extended Release Abuse-Deterrent CSER Capsule Extended Release Sprinkle CEPK Capsule Extended Release Therapy Pack CPCR Capsule Extended Release* CPSP Capsule Sprinkle CPPK Capsule Therapy Pack CART Cartridge CTKT Cartridge Kit CONC Concentrate CREA Cream CRYS Crystals DEVI Device TEST Diagnostic Test DPRH Diaphragm DISK Disk ELIX Elixir EMUL Emulsion ENEM Enema EXHA Exhaler EXHL Exhaler Liquid Dosage Form Description CODE DOSAGE_FORM DOSAGE_FORM DESCRIPTION EXHP Exhaler Powder EXHS Exhaler Solution EXHU Exhaler Suspension FILM Film FLAK Flakes EXTR Fluid Extract FOAM Foam GAS Gas GEL Gel SOLG Gel Forming Solution GRAN Granules GREF Granules Effervescent GUM Gum IMPL Implant INHA Inhaler INJ Injectable INST Insert IUD Intrauterine Device JTAJ Jet-injector (Needleless) JTKT Jet-injector -

Formulation and Evaluation of Liposomal Drug Delivery System for Doxorubicin Hydrochloride
FORMULATION AND EVALUATION OF LIPOSOMAL DRUG DELIVERY SYSTEM FOR DOXORUBICIN HYDROCHLORIDE A dissertation submitted to THE TAMILNADU Dr. M.G.R.MEDICAL UNIVERSITY, CHENNAI. In partial fulfillment of the requirements for the award of degree of MASTER OF PHARMACY IN PHARMACEUTICS BY REG.NO: 26091390 Under the Guidance of Prof. S.P.SENTHIL, M.Pharm., ( Ph.D.,) OCTOBER -2011 THE ERODE COLLEGE OF PHARMACY AND RESEARCH INSTITUTE ERODE -638112, TAMILNADU DEDICATED TO My Beloved Family, Teachers & Friends CERTIFICATES The Erode College Of Pharmacy and Research Institute Prof.S.P.SENTHIL, M.Pharm.,( Ph.D.,) Department of pharmaceutics, Perundurai Main Road, Veppampalayam, Erode-638112, India. e-mail : [email protected] CERTIFICATE This is to certify that the investigation in this thesis entitled “FORMULATION AND EVALUATION OF LIPOSOMAL DRUG DELIVERY SYSTEM FOR DOXORUBICIN HYDROCHLORIDE” submitted to The Tamilnadu Dr. M.G.R. Medical University Chennai. For partial fulfillment of the award of degree of Master of pharmacy in Pharmaceutics was carried out by Reg. No: 26091390 in the department of pharmaceutics, The Erode College of pharmacy, Erode, under my guidance and supervision This work is original and has not been submitted in part or full to any other degree or diploma of this or any other university. Place: Erode Prof. S.P.SENTHIL, M.Pharm.,(Ph.D.,) Date: The Erode College Of Pharmacy and Research Institute Dr.V.Ganesan, M.Pharm., Ph.D., Professor and HOD of Pharmaceutics, Perundurai Main Road, Veppampalayam, Erode-638112, India. CERTIFICATE This is to certify that the investigation in this thesis entitled “FORMULATION AND EVALUATION OF LIPOSOMAL DRUG DELIVERY SYSTEM FOR DOXORUBICIN HYDROCHLORIDE ” submitted to the Tamil Nadu Dr. -

Medicinal Cannabis Dosage Forms in California an Overview of Cannabis Dosage Forms
Mica Gross President Sante Botanica / COO MDBioLogics Medicinal Cannabis Dosage Forms in California An Overview of Cannabis Dosage Forms Forms of dosage are the specific vehicles by which cannabis, or cannabinoids, are delivered into the body. Cannabis (drug-containing plant) —> The “form” in which THC and other cannabinoids Extracted Oil (drug substance) —> are administered determines how they moves and Delivery / Intake (drug product) works medicinally inside the body: ie the pharmacokinetics of cannabis. Both the plant material and the oils yielded through the extraction process influence the performance of the different forms of intake, which each have a particular set of benefits. An Overview of Cannabis Dosage Forms The medicinal effect experienced by the end- user is a function of the quality of the raw material – the cannabinoid and terpene profile of the cannabis – and of the derived oil yielded during extraction. The goal is to preserve the fidelity of the volatile medicinal compounds – such as the cannabinoids, terpenes, and flavonoids – throughout the entire chain of custody so that the therapeutic effect is intact and the dose is delivered in the most efficacious means possible. An Overview of Cannabis Dosage Forms Each form of intake is formulated to take advantage of the pharmacokinetic pathways as effectively as possible in delivering a therapeutic dose of cannabinoids (e.g. THC or CBD). An Overview of Cannabis Dosage Forms Important factors when considering each form of intake: • Bioavailability – the fraction of the administered -

Aqueous Emulsion Coating for Pharmaceutical Dosage Form
Europaisches Patentamt 0 347 024 3> European Patent Office J) Publication number: Dffice europeen des brevets 3) EUROPEAN PATENT APPLICATION © Application number: 89303290.4 © Int. CI.4: A61K 9/32 , A61K 9/36 , A61K 9/22 © Date of filing: 04.04.89 The title of the invention has been amended © Applicant: ALZA CORPORATION (Guidelines for Examination in the EPO, A-lll, 950 Page Mill Road P.O. Box 10950 7.3). Palo Alto California 94303-0802(US) © Inventor: Wong, Patrick S.-L. © Priority: 21.04.88 US 184478 2030 Cornell Street Palo Alto California 94306(US) © Date of publication of application: Inventor: Theeuwes, Felix 20.12.89 Bulletin 89/51 1634 Fallen Leaf Lane Los Altos California 94022(US) © Designated Contracting States: AT BE CH DE ES FR GB GR IT LI LU NL SE © Representative: Evans, David Charles et al F.J. CLEVELAND & COMPANY 40-43, Chancery Lane London, WC2A UQ(GB) © Aqueous emulsion coating for pharmaceutical dosage form. © A dosage form is disclosed comprising a cured emulsion coat that surrounds a drug. The emulsion comprises a lower alkyl acrylate-lower alkyl methacrylate copolymer and ethyl cellulose. The emulsion optionally comprises a hydrophilic poly- mer. eg < CM CO CL LU Xerox Copy Centre 1 EP 0 347 024 A2 2 AQUEOUS EMULSION FOR PHARMACEUTICAL DOSAGE FORM This invention pertains to a pharmaceutical final dosage form. dosage form comprising an aqueous emulsion coat It will be appreciated by those skilled in the and to an aqueous emulsion coat. drug dispensing art that if a coating is provided that is substantially free of organic solvents when coat- 5 ing drugs, drug granules, drug powders, drug dis- BACKGROUND OF THE INVENTION pensers, and the like, such a coating would have an immediate positive value and, concomitantly, represent an advancement in the drug coating art. -

Dosage Form FDA Data Element Number
Home Drugs Development & Approval Process (Drugs) Forms & Submission Requirements Electronic Submissions to CDER Data Standards Manual (monographs) Drugs Dosage Form FDA Data Element Number. None. CDER Data Element Number. C-DRG-00201 Data Element Name. Dosage Form. Data Element OID: 2.16.840.1.113883.3.26.1.1.2 Data Element NCI Concept ID: C42636 Version Number. 008 Description. This standard provides for all drug dosage forms. The granularity of data often requires that more specific dosage form terms be stored in automated databases than are represented in publications. These dosage form terms are available not only for use in databases that track approved drug products, but also for drug products such as: those that have not been approved, investigational drug products, homeopathic drug products, biologic products, veterinary drug products, and bulk drug products. A ‘use restrictions’ column is being added to help identify where a particular dosage form set or subset should be used (a=CDER Databases, b=NDC Directory, c=Orange Book). The definitions for Lotion, Cream, Ointment, and Paste were revised on June 21, 2006 to include information that would assist the user in differentiating between these dosage forms. These changes were the result of discussions during an FDA Advisory Committee with an open public forum, scientific studies that were published in refereed pharmaceutical journals, and internal discussions by Agency personnel, including the usual adherence to the criteria that are specified in MaPP 7600.4. A 1999 Food and Drug Administration Draft Guidance for Industry states: "A dosage form is the way of identifying the drug in its physical form. -

Thin Films As an Emerging Platform for Drug Delivery
View metadata, citation and similar papers at core.ac.uk brought to you by CORE provided by Elsevier - Publisher Connector asian journal of pharmaceutical sciences 11 (2016) 559–574 HOSTED BY Available online at www.sciencedirect.com ScienceDirect journal homepage: www.elsevier.com/locate/ajps Review Thin films as an emerging platform for drug delivery Sandeep Karki a,1, Hyeongmin Kim a,b,c,1, Seon-Jeong Na a, Dohyun Shin a,c, Kanghee Jo a,c, Jaehwi Lee a,b,c,* a Pharmaceutical Formulation Design Laboratory, College of Pharmacy, Chung-Ang University, Seoul 06974, Republic of Korea b Bio-Integration Research Center for Nutra-Pharmaceutical Epigenetics, Chung-Ang University, Seoul 06974, Republic of Korea c Center for Metareceptome Research, Chung-Ang University, Seoul 06974, Republic of Korea ARTICLE INFO ABSTRACT Article history: Pharmaceutical scientists throughout the world are trying to explore thin films as a novel Received 21 April 2016 drug delivery tool. Thin films have been identified as an alternative approach to conven- Accepted 12 May 2016 tional dosage forms. The thin films are considered to be convenient to swallow, self- Available online 6 June 2016 administrable, and fast dissolving dosage form, all of which make it as a versatile platform for drug delivery. This delivery system has been used for both systemic and local action via Keywords: several routes such as oral, buccal, sublingual, ocular, and transdermal routes. The design Thin film of efficient thin films requires a comprehensive knowledge of the pharmacological and phar- Film-forming polymer maceutical properties of drugs and polymers along with an appropriate selection of Mechanical properties manufacturing processes. -

API & Dosage Form Development
API and Dosage Development W I T H D A LT O N Peter Pekos [ C O M P A N Y V I S I O N ] "To make the impossible possible. Dalton Pharma Services uses its scientific and pharmaceutical expertise to bring customer ideas to life. We develop their new drug products, optimize the synthesis of therapeutic candidates, and manufacture them at the highest level of quality." [ S E R V I C E S ] Contract Research Custom Synthesis Medicinal and Flow Chemistry API Process Development Formulation Development cGMP API Manufacturing cGMP Sterile Filling Analytical and Microbiology Services FDA inspected, HC approved, & MRA with EMA API Form Development API Definition Active pharmaceutical ingredient (API) – the substance(s) in pharmaceutical drugs that is/are responsible for the beneficial health effects experienced by consumers. (1) API Selection Key factors that drive API selection (2): Crystallinity: Influences the dissolution rate and transport characteristics of the drug (3) Polymorphism: The ability of a drug substance to Solubility: take on more than one form/ The ability of a solute to dissolve in crystalline phase. Influences drug solvent. Influences desired dissolution and drug stability (i.e., concentration and drug premature degradation) (4) absorption (6) Density: Influences flow properties and Stability: compressibility (5) Influenced by moisture, excipients, temperature, pH, oxygen, light (7) Particle size distribution: Influences the ability of the drug to Density: cross blood barriers, enter cells, Influences flow properties and and absorbed by the human body compressibility (5) (2) The development stage and needs API & CTD Note: In any clinical application or new drug submission the 1) description of manufacturing (including flowcharts) is required along with 2) physiochemical characteristics (melting point, boiling point, denaturation temperature, solubility) (2.3.S.2, 2.3.S.3 and 2.3.P.3) (4) Once the API form is determined, the dosage form must be selected. -

Achievements in Thermosensitive Gelling Systems for Rectal Administration
International Journal of Molecular Sciences Review Achievements in Thermosensitive Gelling Systems for Rectal Administration Maria Bialik, Marzena Kuras, Marcin Sobczak and Ewa Oledzka * Department of Biomaterials Chemistry, Chair of Analytical Chemistry and Biomaterials, Faculty of Pharmacy, Medical University of Warsaw, 1 Banacha St., 02-097 Warsaw, Poland; [email protected] (M.B.); [email protected] (M.K.); [email protected] (M.S.) * Correspondence: [email protected]; Tel.: +48-22-572-07-55 Abstract: Rectal drug delivery is an effective alternative to oral and parenteral treatments. This route allows for both local and systemic drug therapy. Traditional rectal dosage formulations have histori- cally been used for localised treatments, including laxatives, hemorrhoid therapy and antipyretics. However, this form of drug dosage often feels alien and uncomfortable to a patient, encouraging refusal. The limitations of conventional solid suppositories can be overcome by creating a thermosen- sitive liquid suppository. Unfortunately, there are currently only a few studies describing their use in therapy. However, recent trends indicate an increase in the development of this modern therapeutic system. This review introduces a novel rectal drug delivery system with the goal of summarising recent developments in thermosensitive liquid suppositories for analgesic, anticancer, antiemetic, antihypertensive, psychiatric, antiallergic, anaesthetic, antimalarial drugs and insulin. The report also presents the impact of various types of components and their concentration on the properties of this rectal dosage form. Further research into such formulations is certainly needed in order to meet Citation: Bialik, M.; Kuras, M.; the high demand for modern, efficient rectal gelling systems. Continued research and development Sobczak, M.; Oledzka, E.