Pharmacogenetics in Warfarin Therapy
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Enoxaparin Sodium Solution for Injection, Manufacturer's Standard
PRODUCT MONOGRAPH INCLUDING PATIENT MEDICATION INFORMATION PrLOVENOX® Enoxaparin sodium solution for injection 30 mg in 0.3 mL solution (100 mg/mL), pre-filled syringes for subcutaneous or intravenous injection 40 mg in 0.4 mL solution (100 mg/mL), pre-filled syringes for subcutaneous or intravenous injection 60 mg in 0.6 mL solution (100 mg/mL), pre-filled syringes for subcutaneous or intravenous injection 80 mg in 0.8 mL solution (100 mg/mL), pre-filled syringes for subcutaneous or intravenous injection 100 mg in 1 mL solution (100 mg/mL), pre-filled syringes for subcutaneous or intravenous injection 300 mg in 3 mL solution (100 mg/mL), multidose vials for subcutaneous or intravenous injection PrLOVENOX® HP Enoxaparin sodium (High Potency) solution for injection 120 mg in 0.8 mL solution (150 mg/mL), pre-filled syringes for subcutaneous or intravenous injection 150 mg in 1 mL solution (150 mg/mL), pre-filled syringes for subcutaneous or intravenous injection Manufacturer’s standard Anticoagulant/Antithrombotic Agent ATC Code: B01AB05 Product Monograph – LOVENOX (enoxaparin) Page 1 of 113 sanofi-aventis Canada Inc. Date of Initial Approval: 2905 Place Louis-R.-Renaud February 9, 1993 Laval, Quebec H7V 0A3 Date of Revision September 7, 2021 Submission Control Number: 252514 s-a version 15.0 dated September 7, 2021 Product Monograph – LOVENOX (enoxaparin) Page 2 of 113 TABLE OF CONTENTS Sections or subsections that are not applicable at the time of authorization are not listed. TABLE OF CONTENTS .............................................................................................................. -

Rivaroxaban Versus Warfarin for Treatment and Prevention Of
Costa et al. Thrombosis Journal (2020) 18:6 https://doi.org/10.1186/s12959-020-00219-w RESEARCH Open Access Rivaroxaban versus warfarin for treatment and prevention of recurrence of venous thromboembolism in African American patients: a retrospective cohort analysis Olivia S. Costa1,2, Stanley Thompson3, Veronica Ashton4, Michael Palladino5, Thomas J. Bunz6 and Craig I. Coleman1,2* Abstract Background: African Americans are under-represented in trials evaluating oral anticoagulants for the treatment of acute venous thromboembolism (VTE). The aim of this study was to evaluate the effectiveness and safety of rivaroxaban versus warfarin for the treatment of VTE in African Americans. Methods: We utilized Optum® De-Identified Electronic Health Record data from 11/1/2012–9/30/2018. We included African Americans experiencing an acute VTE during a hospital or emergency department visit, who received rivaroxaban or warfarin as their first oral anticoagulant within 7-days of the acute VTE event and had ≥1 provider visit in the prior 12-months. Differences in baseline characteristics between cohorts were adjusted using inverse probability-of-treatment weighting based on propensity scores (standard differences < 0.10 were achieved for all covariates). Our primary endpoint was the composite of recurrent VTE or major bleeding at 6-months. Three- and 12-month timepoints were also assessed. Secondary endpoints included recurrent VTE and major bleeding as individual endpoints. Cohort risk was compared using Cox regression and reported as hazard ratios (HRs) with 95% confidence intervals (CIs). Results: We identified 2097 rivaroxaban and 2842 warfarin users with incident VTE. At 6-months, no significant differences in the composite endpoint (HR = 0.96, 95%CI = 0.75–1.24), recurrent VTE (HR = 1.02, 95%CI = 0.76–1.36) or major bleeding alone (HR = 0.93, 95%CI = 0.59–1.47) were observed between cohorts. -

Pharmacokinetics of Anticoagulant Rodenticides in Target and Non-Target Organisms Katherine Horak U.S
University of Nebraska - Lincoln DigitalCommons@University of Nebraska - Lincoln USDA National Wildlife Research Center - Staff U.S. Department of Agriculture: Animal and Plant Publications Health Inspection Service 2018 Pharmacokinetics of Anticoagulant Rodenticides in Target and Non-target Organisms Katherine Horak U.S. Department of Agriculture, [email protected] Penny M. Fisher Landcare Research Brian M. Hopkins Landcare Research Follow this and additional works at: https://digitalcommons.unl.edu/icwdm_usdanwrc Part of the Life Sciences Commons Horak, Katherine; Fisher, Penny M.; and Hopkins, Brian M., "Pharmacokinetics of Anticoagulant Rodenticides in Target and Non- target Organisms" (2018). USDA National Wildlife Research Center - Staff Publications. 2091. https://digitalcommons.unl.edu/icwdm_usdanwrc/2091 This Article is brought to you for free and open access by the U.S. Department of Agriculture: Animal and Plant Health Inspection Service at DigitalCommons@University of Nebraska - Lincoln. It has been accepted for inclusion in USDA National Wildlife Research Center - Staff ubP lications by an authorized administrator of DigitalCommons@University of Nebraska - Lincoln. Chapter 4 Pharmacokinetics of Anticoagulant Rodenticides in Target and Non-target Organisms Katherine E. Horak, Penny M. Fisher, and Brian Hopkins 1 Introduction The concentration of a compound at the site of action is a determinant of its toxicity. This principle is affected by a variety of factors including the chemical properties of the compound (pKa, lipophilicity, molecular size), receptor binding affinity, route of exposure, and physiological properties of the organism. Many compounds have to undergo chemical changes, biotransformation, into more toxic or less toxic forms. Because of all of these variables, predicting toxic effects and performing risk assess- ments of compounds based solely on dose are less accurate than those that include data on absorption, distribution, metabolism (biotransformation), and excretion of the compound. -

Warfarin in Antiphospholipid Syndrome — Time to Explore New Horizons
Editorial Warfarin in Antiphospholipid Syndrome — Time to Explore New Horizons Antiphospholipid syndrome (APS) constitutes vascular boembolism have significant reduction in the risk of recur- thrombosis and/or pregnancy morbidity occurring in per- rent venous thrombosis when they continue to receive war- sons with antiphospholipid antibodies (aPL), most com- farin with a target international normalized ratio (INR) of monly a positive lupus anticoagulant (LAC) test, anticardi- 1.5 to 2.019. The risk of venous thromboembolism is rough- ly 10% per year whether warfarin is stopped after 3, 6, 12, olipin antibodies (aCL), and anti-ß2-glycoprotein I antibod- 1 or 27 months in aPL-negative patients with unprovoked ies (ß2-GPI) . Given the wide spectrum of aPL-related clin- ical manifestations and significant morbidity and mortality thrombosis20. due to APS2, primary and secondary thrombosis prevention Thus, no compelling data exist about the optimal dura- is crucial. Primary thrombosis prevention lacks an evidence- tion of therapy or when anticoagulation can be discontinued based approach; controlled, prospective, and randomized in APS patients. Ideally, anticoagulation should be stopped studies are in progress3,4. For secondary thrombosis preven- when the risks of treatment outweigh the risks of thrombo- tion, the current recommendation is life-long warfarin; the sis21. Currently it is not possible to predict which patients necessity, duration, and intensity of warfarin treatment are with APS will develop recurrent thrombosis when warfarin still under debate. Further, warfarin use is cumbersome due treatment is stopped, and there is no evidence for or against to bleeding complications, frequent blood monitoring, and indefinite anticoagulation in patients with APS who devel- teratogenicity. -

Warfarin I G Y H H O S I - L W T U A
Read this important information before taking: Warfarin Brought to you by the Institute for Safe Medication Practices [ Extra care is needed because warfarin is a high-alert medicine. ] High-alert medicines have been proven to be safe and effective. But these medicines can cause serious injury if a mistake happens while taking them. This means that it is very important for you to know about this medicine and take it exactly as directed. When taking warfarin (blood thinner) 1 Take exactly as directed. Take your medicine at the same time each day. Do not take extra doses or skip any doses. n i When the doctor changes your dose r a 2 Keep a record of telephone calls. f When your doctor, nurse, or pharmacist calls to r change your dose: write down the dose and any other instructions; read the dose and in - a structions back to him or her to make sure you understand them; and date the instructions W so they won’t be mixed up with older instructions. r o f 3 A D M I N Know your dose. U Always tell your doctor the strength of warfarin tablets that you have on O U M A D C O I N C s hand. Then ask him or her how much warfarin to take, and how many tablets in that strength p i to take to equal the dose. If you are running low on tablets, ask for a new prescription. T y 4 t Keep instructions nearby. Keep the dated instructions near the medicine, and read RX e f them every time before taking your warfarin. -

Electrochemical Synthesis of Novel 1,3-Indandione Derivatives and Evaluation of Their Antiplatelet Aggregation Activities
Iranian Journal of Pharmaceutical Research (2013), 12 (supplement): 91-103 Copyright © 2013 by School of Pharmacy Received: September 2012 Shaheed Beheshti University of Medical Sciences and Health Services Accepted: February 2013 Original Article Electrochemical Synthesis of Novel 1,3-Indandione Derivatives and Evaluation of Their Antiplatelet Aggregation Activities Salimeh Amidia, Farzad Kobarfardab*, Abdolmajid Bayandori Moghaddamc Kimia Tabiba and Zohreh Soleymania aDepartment of Medicinal Chemistry, School of Pharmacy, Shahid Beheshti University of Medical Sciences, Tehran, Iran. bPhytochemistry Research Center, Shahid Beheshti University of Medical Sciences, Tehran, Iran. cDepartment of Engineering Science, College of Engineering, University of Tehran, P.O. Box 11155-4563, Tehran, Iran. Abstract Electrochemical oxidation of some selected catechol derivatives, using cyclic voltammetry, in the presence of different 2-aryl-1,3-indandiones as nucleophiles, resulted in electrochemical synthesis of new 1,3- indandione derivatives in an undivided cell in good yield and purity. A Michael addition mechanism was proposed for the formation of the analogs based on the reaction conditions which were provided in electrochemical cell. The in-vitro antiplatelet and anticoagulant activity of these compounds was evaluated, using arachidonic acid (AA) and adenosine diphosphate (ADP) as the platelet aggregation inducers. The results show that the incorporation of catechol ring in 1,3-indandione nucleus leads to the emergence of antiplatelet aggregation activity in these compounds. The compounds may exert their antiaggregation activity by interfering with the arachidonic acid pathway. Keywords:Electrochemical synthesis; 1,3-Indandione; Platelet aggregation inhibition. Introduction anticoagulants and platelet aggregation inhibitors is frequently used in high risk patients in order Stroke and cardiovascular diseases are the to achieve more efficient clinical results (4-5). -

Risk of Bleeding with Single, Dual, Or Triple Therapy with Warfarin, Aspirin, and Clopidogrel in Patients with Atrial Fibrillation
ORIGINAL INVESTIGATION LESS IS MORE Risk of Bleeding With Single, Dual, or Triple Therapy With Warfarin, Aspirin, and Clopidogrel in Patients With Atrial Fibrillation Morten L. Hansen, MD, PhD; Rikke Sørensen, MD; Mette T. Clausen, MSc Pharm; Marie Louise Fog-Petersen, MSc Pharm; Jakob Raunsø, MD; Niels Gadsbøll, MD, DMSc; Gunnar H. Gislason, MD, PhD; Fredrik Folke, MD; Søren S. Andersen, MD; Tina K. Schramm, MD; Steen Z. Abildstrøm, MD, PhD; Henrik E. Poulsen, MD, DMSc; Lars Køber, MD, DMSc; Christian Torp-Pedersen, MD, DMSc Background: Patients with atrial fibrillation (AF) of- est for dual clopidogrel and warfarin therapy (13.9% per ten require anticoagulation and platelet inhibition, but patient-year) and triple therapy (15.7% per patient- data are limited on the bleeding risk of combination year). Using warfarin monotherapy as a reference, the therapy. hazard ratio (95% confidence interval) for the com- bined end point was 0.93 (0.88-0.98) for aspirin, 1.06 Methods: We performed a cohort study using nation- (0.87-1.29) for clopidogrel, 1.66 (1.34-2.04) for aspirin- wide registries to identify all Danish patients surviving clopidogrel, 1.83 (1.72-1.96) for warfarin-aspirin, 3.08 first-time hospitalization for AF between January 1, 1997, (2.32-3.91) for warfarin-clopidogrel, and 3.70 (2.89- and December 31, 2006, and their posthospital therapy 4.76) for warfarin-aspirin-clopidogrel. of warfarin, aspirin, clopidogrel, and combinations of these drugs. Cox proportional hazards models were used to es- Conclusions: In patients with AF, all combinations of timate risks of nonfatal and fatal bleeding. -

Understanding Warfarin (Coumadin) Brochure
Phone number Appointment date I willfollowupwith Notes Date: Warfarin (Coumadin) mg tablets INR: Sun Mon Tues Wed Thurs Fri Sat REV 7/12J14849DC14120 Tab(s) Tab(s) Tab(s) Tab(s) Tab(s) Tab(s) Tab(s) (Coumadin®) Warfarin Understanding Key Questions What is Warfarin? 1. Why am I taking warfarin (Coumadin®, Your doctor wants you to take the drug Jantoven®)? warfarin. Warfarin is the generic name – To keep from getting a blood clot for this drug. You may have also heard it called Coumadin® or Jantoven®. 2. What do I need to do to take this medication safely? Warfarin can help stop blood clots. – Take my medicine at the same time It can also keep clots from getting each day larger. Sometimes blood clots form – Watch for signs of bleeding on surfaces of blood vessels due to – Make sure everyone knows I am taking the drug warfarin damage to that blood vessel. Others form in the heart due to damage in – Keep an up-to-date medication list the heart. When blood clots form, they – Keep my diet steady can break off and move to other parts – Have my blood tested regularly of your body such as your arms, legs, 3. Why is it important to take warfarin lungs and even your brain. If a blood safely? clot blocks a blood vessel, it can stop – To keep from getting a blood clot the flow of blood, damage tissue, and and reduce side effects can even cause death. This brochure will tell you what to expect when you take the drug warfarin. -
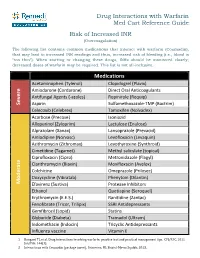
Drug Interactions with Warfarin Med Cart Reference Guide Risk of Increased INR (Overcoagulation)
Drug Interactions with Warfarin Med Cart Reference Guide Risk of Increased INR (Overcoagulation) The following list contains common medications that interact with warfarin (Coumadin), that may lead to increased INR readings and thus, increased risk of bleeding (i.e., blood is “too thin”). When starting or changing these drugs, INRs should be monitored closely; decreased doses of warfarin may be required. This list is not all-inclusive. Medications Acetaminophen (Tylenol) Clopidogrel (Plavix) Amiodarone (Cordarone) Direct Oral Anticoagulants Antifungal Agents (‐azoles) Ropinirole (Requip) Severe Aspirin Sulfamethoxazole‐TMP (Bactrim) Celecoxib (Celebrex) Tamoxifen (Nolvadex) Acarbose (Precose) Isoniazid Allopurinol (Zyloprim) Lactulose (Enulose) Alprazolam (Xanax) Lansoprazole (Prevacid) Amlodipine (Norvasc) Levofloxacin (Levaquin) Azithromycin (Zithromax) Levothyroxine (Synthroid) Cimetidine (Tagamet) Methyl salicylate (topical) Ciprofloxacin (Cipro) Metronidazole (Flagyl) Clarithromycin (Biaxin) Moxifloxacin (Avelox) Colchicine Omeprazole (Prilosec) Doxycycline (Vibratab) Phenytoin (Dilantin) Moderate Efavirenz (Sustiva) Protease Inhibitors Ethanol Quetiapine (Seroquel) Erythromycin (E.E.S.) Ranitidine (Zantac) Fenofibrate (Tricor, Trilipix) SSRI Antidepressants Gemfibrozil (Lopid) Statins Glyburide (Diabeta) Tramadol (Ultram) Indomethacin (Indocin) Tricyclic Antidepressants Influenza vaccine Vitamin E 1. Bungard TJ, et al. Drug interactions involving warfarin: practice tool and practical management tips. CPJ/RPC. 2011 Jan/Feb. 144(1). 2. Interactions with Coumadin [package insert]. Princeton, NJ: Bristol‐Myers Squibb; 2013. Drug Interactions with Warfarin Med Cart Reference Guide Risk of Decreased INR (Undercoagulation) The following list contains common medications that interact with warfarin (Coumadin), which may lead to decreased INR readings and thus, increased risk of clotting, strokes, and DVTs (i.e., blood is “not thin enough”). When starting or changing these drugs, INRs should be monitored closely; increased doses of warfarin may be required. -

Potential Risks of Nine Rodenticides to Birds and Nontarget Mammals: a Comparative Approach
UNITED STATES ENVIRONMENTAL PROTECTION AGENCY WASHINGTON, D.C. 20460 OFFICE OF PREVENTION, PESTICIDES AND TOXIC SUBSTANCES Potential Risks of Nine Rodenticides to Birds and Nontarget Mammals: a Comparative Approach July 2004 Office of Pesticides Programs Environmental Fate and Effects Division Prepared by: William Erickson, Biologist Environmental Risk Branch 2 Douglas Urban, Senior Biologist Environmental Risk Branch 3 Executive Summary This document presents the Agency’s assessment of potential risks to birds and nontarget mammals from 9 rodenticides, including 3 second-generation anticoagulants (brodifacoum, difethialone, bromadiolone), 3 first-generation anticoagulants (diphacinone, chlorophacinone, warfarin), and 3 non-anticoagulant compounds (zinc phosphide, bromethalin, cholecalciferol). These rodenticides are predominantly used to control commensal rats and mice in and around buildings, transport vehicles, and in sewers. Some, mainly zinc phosphide, chlorophacinone, and diphacinone, also have products registered for other outdoor uses against other rodent and small mammalian pests. A major concern in using rodenticides is that they are not selective to the target species; birds and nontarget mammals that feed on grain-based baits (pellets, meal, treated grains, wax blocks) or meat-based, vegetable, or fruit baits are potentially at risk. The available information from laboratory and pen studies, field studies, control programs, reported incidents, and toxicokinetics also indicates that a variety of avian and mammalian predators and scavengers are potentially at risk from consuming animals poisoned with some of these rodenticides. The assessment focuses on the potential primary and secondary risks to birds and nontarget mammals posed by applications of these 9 rodenticides (11 baits) to control rats and mice in and around buildings (commensal use) and in field and other outdoor settings to control various rodent and other small mammalian pests. -
Clexane (Enoxaparin) Information for Patients Clexane (Enoxoparin) Information for Patients
Clexane (enoxaparin) Information for patients Clexane (enoxoparin) Information for patients Clexane (enoxaparin) is a type of anticoagulant (blood thinner) that can be used in place of, or in addition to warfarin in certain circumstances. It is given as an injection under the skin, similar to insulin for diabetes. This brochure outlines some things you might need to know about Clexane. How does it work? Clexane is very different to warfarin in the way it works, although it does a similar job. Clexane stops your blood from clotting by deactivating one of the proteins in your blood that your body uses to form a clot. The blood test you would normally use to monitor warfarin (the INR) is not affected by Clexane. The required dose is not based on blood levels, but rather on your size and kidney function. The major advantage of Clexane is that it acts quickly and leaves your body quickly compared to warfarin which acts slowly and leaves your body slowly. When do I need Clexane? Clexane is used when the risk of having a blood clot is very high and the INR is below the therapeutic (target) range. Temporarily using Clexane as treatment in place of warfarin is commonly known as ‘bridging’. There are three common reasons why your doctor might use a Clexane ‘bridge’. 1. You have a high risk of clotting and you are just starting or restarting warfarin. The most common examples include a recent blood clot (Deep Vein Thrombosis (DVT) or pulmonary embolism) or you have a mechanical heart valve. In this case, Clexane is used at the same time as warfarin until the INR is within therapeutic range. -

Anticoagulant Residues in Rats and Secondary Non-Target Risk
Anticoagulant residues in rats and secondary non-target risk DOC SCIENCE INTERNAL SERIES 188 P. Fisher, C. O’Connor, G. Wright and C.T. Eason Published by Department of Conservation PO Box 10-420 Wellington, New Zealand DOC Science Internal Series is a published record of scientific research carried out, or advice given, by Department of Conservation staff or external contractors funded by DOC. It comprises reports and short communications that are peer-reviewed. Individual contributions to the series are first released on the departmental website in pdf form. Hardcopy is printed, bound, and distributed at regular intervals. Titles are also listed in the DOC Science Publishing catalogue on the website, refer http://www.doc.govt.nz under Publications, then Science and Research. © Copyright September 2004, New Zealand Department of Conservation ISSN 1175–6519 ISBN 0–478–22607–1 In the interest of forest conservation, DOC Science Publishing supports paperless electronic publishing. When printing, recycled paper is used wherever possible. This report was prepared for publication by DOC Science Publishing, Science & Research Unit; editing by Katrina Rainey and Helen O’Leary and layout by Geoff Gregory. Publication was approved by the Manager, Science & Research Unit, Science Technology and Information Services, Department of Conservation, Wellington. CONTENTS Abstract 5 1. Introduction 6 1.1 Objectives 6 1.2 Assessing secondary non-target risk 6 2. Methods 8 2.1 Animal husbandry 8 2.2 Feeding trials and analysis of bait samples 8 2.3 Rats offered a lethal amount of bait over 4 days (Trial 1) 10 2.4 One day’s feeding ad libitum on bait (Trial 2) 11 2.5 Ad libitum feeding on a choice of bait and non-toxic pellets until death (Trial 3) 11 2.6 Analysis of tissue samples 12 2.7 Calculation and comparison of potential secondary poisoning risk 12 3.