Supporting Information Structurally
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Energy-Saving Reduced-Pressure Extractive Distillation with Heat
Energy-Saving Reduced-Pressure Extractive Distillation with Heat Integration for Separating the Biazeotropic Ternary Mixture Tetrahydrofuran–Methanol–Water Jinglian Gu, Xinqiang You, Changyuan Tao, Jun Li, Vincent Gerbaud To cite this version: Jinglian Gu, Xinqiang You, Changyuan Tao, Jun Li, Vincent Gerbaud. Energy-Saving Reduced- Pressure Extractive Distillation with Heat Integration for Separating the Biazeotropic Ternary Mixture Tetrahydrofuran–Methanol–Water. Industrial and engineering chemistry research, American Chemical Society, 2018, 57 (40), pp.13498-13510. 10.1021/acs.iecr.8b03123. hal-01957130 HAL Id: hal-01957130 https://hal.archives-ouvertes.fr/hal-01957130 Submitted on 17 Dec 2018 HAL is a multi-disciplinary open access L’archive ouverte pluridisciplinaire HAL, est archive for the deposit and dissemination of sci- destinée au dépôt et à la diffusion de documents entific research documents, whether they are pub- scientifiques de niveau recherche, publiés ou non, lished or not. The documents may come from émanant des établissements d’enseignement et de teaching and research institutions in France or recherche français ou étrangers, des laboratoires abroad, or from public or private research centers. publics ou privés. OATAO is an open access repository that collects the work of Toulouse researchers and makes it freely available over the web where possible This is an author’s version published in: http://oatao.univ-toulouse.fr/21066 Official URL: https://doi.org/10.1021/acs.iecr.8b03123 To cite this version: Gu, Jinglian and You, Xinqiang and Tao, Changyuan and Li, Jun and Gerbaud, Vincent Energy-Saving Reduced-Pressure Extractive Distillation with Heat Integration for Separating the Biazeotropic Ternary Mixture Tetrahydrofuran–Methanol–Water. -

Poly(Ethylene Oxide-Co-Tetrahydrofuran) and Poly(Propylene Oxide-Co-Tetrahydrofuran): Synthesis and Thermal Degradation
Revue Roumaine de Chimie, 2006, 51(7-8), 781–793 Dedicated to the memory of Professor Mircea D. Banciu (1941–2005) POLY(ETHYLENE OXIDE-CO-TETRAHYDROFURAN) AND POLY(PROPYLENE OXIDE-CO-TETRAHYDROFURAN): SYNTHESIS AND THERMAL DEGRADATION Thomas HÖVETBORN, Markus HÖLSCHER, Helmut KEUL∗ and Hartwig HÖCKER Lehrstuhl für Textilchemie und Makromolekulare Chemie der Rheinisch-Westfälischen Technischen Hochschule Aachen, Pauwelsstr. 8, 52056 Aachen, Germany Received January 12, 2006 Copolymers of tetrahydrofuran (THF) and ethylene oxide (EO) (poly(THF-co-EO) and THF and propylene oxide (PO) (poly(THF-co-PO) were obtained by cationic ring opening polymerization of the monomer mixture at 0°C using boron trifluoride etherate (BF3.OEt2) as the initiator. From time conversion plots it was concluded that both monomers are consumed from the very beginning of the reaction and random copolymers are obtained. For poly(THF-co-PO) the molar ratio of repeating units was varied from [THF]/[PO] = 1 to 10; the molar ratio of monomers in the feed corresponds to the molar ratio of repeating units in the copolymer. Thermogravimetric analysis of the copolymers revealed that both poly(THF-co-EO) and poly(THF-co-PO) decompose by ca. 50°C lower than poly(THF) and by ca. 100°C lower than poly(EO); 50% mass loss is obtained at T50 = 375°C for poly(EO), T50 = 330°C for poly(THF) and at T50 = 280°C for both copolymers. The [THF]/[PO] ratio does not influence the decomposition temperature significantly as well. For the copolymers the activation energies of the thermal decomposition (Ea) were determined experimentally from TGA measurements and by density functional calculations on model compounds on the B3LYP/6-31+G* level of theory. -

Tetrahydrofuran
SOME CHEMICALS THAT CAUSE TUMOURS OF THE URINARY TRACT IN RODENTS VOLUME 119 This publication represents the views and expert opinions of an IARC Working Group on the Evaluation of Carcinogenic Risks to Humans, which met in Lyon, 6–13 June 2017 LYON, FRANCE - 2019 IARC MONOGRAPHS ON THE EVALUATION OF CARCINOGENIC RISKS TO HUMANS TETRAHYDROFURAN 1. Exposure Data Boiling point: 65–66 °C (EPA, 2012) Melting point: −108.44 °C (ECHA, 2018) 1.1 Identification of the agent Relative density: 0.883 at 25 °C (water, 1) (ECHA, 2018) 1.1.1 Nomenclature Solubility: Miscible in water (ECHA, 2018) Chem. Abstr. Serv. Reg. No.: 109-99-9 Volatility: Vapour pressure, 19.3 kPa at 20 °C (IPCS, 1997) EC/List No.: 203-726-8 Relative vapour density: 2.5 (air = 1); relative Chem. Abstr. Serv. name: Tetrahydrofuran density of the vapour/air mixture at 20 °C IUPAC systematic name: Oxolane (air = 1): 1.28 (IPCS, 1997) Synonyms: Butane alpha,delta-oxide; butane, Stability: Tetrahydrofuran is prone to oxida- 1,4-epoxy-; cyclotetramethylene oxide; dieth- tion to peroxides, butyric acid, butyralde- ylene oxide; 1,4-epoxybutane; furan, tetra- hyde, and related compounds, mainly on hydro-; furanidine; hydrofuran; oxacyclo- ageing and in the presence of light, heat, pentane; tetramethylene oxide; THF and moisture. The formation of peroxides can be retarded by adding stabilizers such as 1.1.2 Structural and molecular formulae, and hydroquinone or 2,6-di-tert-butyl-p-cresol at relative molecular mass 250 mg/kg (Coetzee & Chang, 1985; Müller, 2012). O Flash point: −14.5 °C -

United States Patent 0 Ice Patented Jan
3,489,528 United States Patent 0 ice Patented Jan. 13, 1970 1 2 number of (BHZNHZ) groups, but the product is con 3,489,528 PREPARATION OF POLYAMINOBORANES sistent in composition, as shown by infra-red and ele William E. Zanieski, Pittsburgh, Pa., assignor to Mine mental analyses, and the average value of n is about 4. Safety Appliances Company, a corporation of In another example a solution of THF-EH3 contain— Pennsylvania ing 2.99 g. of B2H6 in 150 ml. of THF was prepared N0 Drawing. Filed Apr. 5, 1965, Ser. No. 445,707 in the same manner as the previous example and the Int. Cl. C01b 21/00 solution was warmed to -30° C. 3.12 g. of gaseous US. Cl. 23—358 6 Claims ammonia was bubbled into and dissolved in the solution at —30° C. and the resultant clear solution was warmed to 0° C, at which temperature hydrogen was evolved and polyaminoborane, identical to that obtained in the ABSTRACT OF THE DISCLOSURE previous example, precipitated from the reaction mix Ammonia and diborane are reacted in tetrahydrofuran ture. at a temperature below about —30° C.; polyaminoboranes ‘Although the invention is not limited to any particular are precipitated when the reaction mixture is warmed. 15 reaction mechanism, it appears that the reaction of NH3 Aging of an ammonia solution of the polyaminoboranes and BZHS in THF forms an intermediate that is formed so produced yields an ammonia insoluble, more highly and is stable only at temperatures below about —30° C. polymerized polyaminoborane. and that this intermediate decomposes at temperatures above about —30° C. -

Tetrahydrofuran (THF)
Tetrahydrofuran (THF) Typical Physical Properties of THF Molecular Weight 72.11 g/mol Empirical Formula C4H8O Appearance Colorless Freezing Point ‐108°C (‐ 164.4°F) Flash Point – Closed Cup ‐20°C (‐4°F) Boiling Point @ 760mmHg 66°C (150.8°F) Autoignition Temperature 230°C (446°F) Density @ 20°C 0.89 kg/l Tetrahydrofuran (THF) is a clear, colorless liquid with 7.43 lb/gal an ether‐like odor. The principal end uses of THF Vapor Pressure @ 20°C 193 hPa include PTMEG, pharmaceutical solvent, adhesives, PVC cement and magnetic tape. Due to its' tendency Evaporation Rate (nBuAc = 1) 8 to form peroxides during storage, THF is inhibited with Solubility @ 20°C (in Water) Complete BHT. Refractive Index @ 20°C 1.407 CAS number Viscosity @ 20°C 0.516 cP Lower Flammability in Air 1.5% v/v 109‐99‐9 Upper Flammability in Air 12.4% v/v Synonyms Note: The properties reported above are typical physical properties. Monument Chemical in no way guarantees that the 1,4‐epoxybutane; Cyclotetramethylene oxide; Butane product from any particular lot will conform exactly to the given .alpha.,.delta.‐oxide values. Health and safety information Under current U.S. OSHA's Hazardous Communication program THF is classified as a flammable liquid, can cause eye and respiratory irritation. Keep the material away from heat sources, hot surfaces, open flames, and sparks. Use only in a well‐ventilated area. Observe good industrial hygiene practices and use appropriate Personal Protective Equipment. For full safety information please refer to the Safety Data Sheet. Storage and Handling specific product Technical Data Sheet (TDS) is accurate, but makes no representations as to the sufficiency or THF should be stored only in tightly closed, properly completeness of the product or information in this TDS vented containers away from heat, sparks, open flame and makes no commitment to correct or bring up‐to‐date or strong oxidizing agents. -

Contribution of Cyclopentyl Methyl Ether (CPME) to Green Chemistry
sustainability / green chemistry Industry perspective Shunji Sakamoto Contribution of Cyclopentyl Methyl Ether (CPME) to Green Chemistry Shunji sakamoto Specialty Chemicals Division, ZEON CORPORATION, 1-6-2 Marunouchi, Chiyoda-ku, Tokyo, 1008246, Japan easy peroxide formation and water miscibility, the demand KEyWORDS for CPME has steadily been growing in the pharmaceutical Cyclopentyl methyl ether, CPME, Green Chemistry, industry as well as in the electronic and fragrance solvent, process, ICH. industries. Also, CPME has been registered or listed in the corresponding legislations for new chemical substances of United States of America, Canada, European Union, ABSTRACT Japan, South Korea, China and Chinese Taipei, and is commercially available in these countries and regions as Cyclopentyl methyl ether (CPME) is an alternative well as others including India. solvent and has preferable properties including higher In this article, I answer the question why CPME is “Green” hydrophobicity, lower formation of peroxides and better even though it is derived from petroleum. stability under acidic and basic conditions compared to other traditional ether solvents such as tetrahydrofuran. Due to the above unique properties, CPME has particular UNIQUE FEATURES OF CPME 24 advantages and renders some conventional reaction sequences in one pot or easier process as a result of CPME is an asymmetric aliphatic ether having a cyclopentyl solvent unification and facile isolation of the products, group. It has seven unique features attributed to its structure which contribute to Green Chemistry as well as to (Table 1). process innovation. CPME meets eight definitions out of the 12 Principles of Green Chemistry (1) because of Extremely lower miscibility with water the above characteristics as well as its manufacturing The solubility of CPME in water is only 1.1% and that of water process and its applications. -

Q3C (R8): Impurities: Guideline for Residual Solvents Step 2B
04 May 2020 EMA/CHMP/ICH/213867/2020 Committee for Medicinal Products for Human Use Q3C (R8): Impurities: guideline for residual solvents Step 2b Transmission to CHMP 30 April 2020 Adoption by CHMP 30 April 2020 Release for public consultation 4 May 2020 Deadline for comments 30 July 2020 Comments should be provided using this template. The completed comments form should be sent to [email protected] Official address Domenico Scarlattilaan 6 ● 1083 HS Amsterdam ● The Netherlands Address for visits and deliveries Refer to www.ema.europa.eu/how-to-find-us Send us a question Go to www.ema.europa.eu/contact Telephone +31 (0)88 781 6000An agency of the European Union © European Medicines Agency, 2020. Reproduction is authorised provided the source is acknowledged. PDE for 2-Methyltetrahydrofuran, Cyclopentyl Methyl Ether, and Tertiary-Butyl Alcohol INTERNATIONAL COUNCIL FOR HARMONISATION OF TECHNICAL REQUIREMENTS FOR PHARMACEUTICALS FOR HUMAN USE ICH HARMONISED GUIDELINE IMPURITIES: GUIDELINE FOR RESIDUAL SOLVENTS Q3C(R8) PDE FOR 2-METHYLTETRAHYDROFURAN, CYCLOPENTYL METHYL ETHER, AND TERTIARY-BUTYL ALCOHOL Draft version Endorsed on 25 March 2020 Currently under public consultation Note : This document contains only the PDE levels for three solvents: 2- methyltetrahydrofuran, cyclopentylmethylether and tert-butanol that were agreed to be included in the ICH Q3C(R8) revision. Further to reaching Step 4, these PDEs would be integrated into a complete Q3C(R8) Guideline document. At Step 2 of the ICH Process, a consensus draft text or guideline, agreed by the appropriate ICH Expert Working Group, is transmitted by the ICH Assembly to the regulatory authorities PDE for 2-Methyltetrahydrofuran, Cyclopentyl Methyl Ether, and Tertiary-Butyl Alcohol of the ICH regions for internal and external consultation, according to national or regional procedures. -

1- MANUFACTURING of MATERIAL BASED HYDROGEN FUEL for LIGHTWEIGHT VEHICLES by ALI KHAGHANI a Thesis
MANUFACTURING OF MATERIAL BASED HYDROGEN FUEL FOR LIGHTWEIGHT VEHICLES Item Type text; Electronic Thesis Authors KHAGHANI, ALI Publisher The University of Arizona. Rights Copyright © is held by the author. Digital access to this material is made possible by the University Libraries, University of Arizona. Further transmission, reproduction or presentation (such as public display or performance) of protected items is prohibited except with permission of the author. Download date 23/09/2021 12:36:34 Link to Item http://hdl.handle.net/10150/613155 MANUFACTURING OF MATERIAL BASED HYDROGEN FUEL FOR LIGHTWEIGHT VEHICLES By ALI KHAGHANI ____________________ A Thesis Submitted to The Honors College In Partial Fulfillment of the Bachelors degree With Honors in Chemical Engineering THE UNIVERSITY OF ARIZONA M A Y 2016 Approved by: ____________________________ Dr. Kimberly Ogden Chemical and Environmental Engineering -1- Abstract Vehicles powered by hydrogen fuel cells store hydrogen as a cooled liquid at 20 degrees kelvin or a compressed gas at 10,000 pounds per square inch. An alternative that eliminates the need for these extremes of temperature and pressure is to heat a compound containing covalently bonded hydrogen, causing it to release the hydrogen to the fuel cell. Ammonia borane, which is stable at ambient conditions, requires minimal energy for dehydrogenation, and is rich in hydrogen, is a possible storage medium for hydrogen. If a viable storage system could be engineered, demand for ammonia borane as a source of hydrogen would increase. The goal of this project is to develop a processing plant and to optimize design specifications for scaling up processing of ammonia borane through the metathesis reaction pathway. -
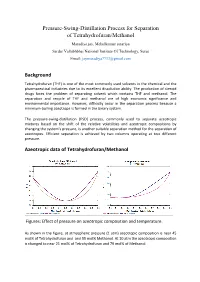
Pressure-Swing-Distillation Process for Separation of Tetrahydrofuran/Methanol
Pressure-Swing-Distillation Process for Separation of Tetrahydrofuran/Methanol Maradiya jay, Mehulkumar sutariya Sardar Vallabhbhai National Institute Of Technology, Surat Email: [email protected] Background Tetrahydrofuran (THF) is one of the most commonly used solvents in the chemical and the pharmaceutical industries due to its excellent dissolution ability. The production of steroid drugs faces the problem of separating solvent which contains THF and methanol. The separation and recycle of THF and methanol are of high economic significance and environmental importance. However, difficulty occur in the separation process because a minimum-boiling azeotrope is formed in the binary system. The pressure-swing-distillation (PSD) process, commonly used to separate azeotropic mixtures based on the shift of the relative volatilities and azeotropic compositions by changing the system’s pressure, is another suitable separation method for the separation of azeotropes. Efficient separation is achieved by two columns operating at two different pressure. Azeotropic data of Tetrahydrofuran/Methanol Figures: Effect of pressure on azeotropic composition and temperature. As shown in the figure, at atmospheric pressure (1 atm) azeotropic composition is near 45 mol% of Tetrahydrofuran and and 55 mol% Methanol. At 10 atm the azeotropic composition is changed to near 21 mol% of Tetrahydrofuran and 79 mol% of Methanol. Description of Flow-Sheet In this flow-sheet We use two distillation column with different pressure (1 atm and 10 atm). In low pressure column we enter a feed; This feed are concentrated near the azeotropic composition at 1 atm,and the 99.9 mol% Methanol are collected from the bottom. The top stream of low pressure column are now enter to the high pressure column where it’s composition are changed with the azeotropic composition at 10 atm, and the pure Tetrahydrofuran are collected from the bottom of that column and the top stream of high pressure column are recycled to the low pressure column. -

Material Safety Data Sheet
MATERIAL SAFETY DATA SHEET MSDS Number: 1201E Section 1 PRODUCT AND COMPANY IDENTIFICATION Trade Name: OATEY CPVC MEDIUM ORANGE CEMENT Product Nos.: 31127, 31128(TV), 31129(TV), 31130, 31131, 32212, 32213, 32214, 32215, 31151, 30234 Product Use: Cement for CPVC Plastic Pipe Formula: CPVC Resin in Solvent Solution Synonyms: CPVC Plastic Pipe Cement Firm Name & Oatey Company 4700 West 160th Street, Cleveland, Ohio 44135 Address: www.oatey.com Firm Phone No: (216) 267-7100 Emergency Phone For Emergency First Aid call 1-877-740-5015. For chemical transportation Nos.: emergencies ONLY, call Chemtrec at 1-800-424-9300. Outside the U.S. 1- 703-527-3887. Prepared by: Technical Department Preparation Date: 07/30/2012 Section 2 HAZARDS IDENTIFICATION Emergency Overview: Orange liquid with an ether-like odor. Extremely flammable liquid and vapor. Vapors may cause flash fire. May cause eye and skin irritation. Inhalation of vapors or mist may cause respiratory irritation and central nervous system effects. Swallowing may cause irritation, nausea, vomiting, diarrhea and kidney or liver disorders. Aspiration hazard. May be fatal if swallowed. Symptoms may be delayed. Section 3 COMPOSITION/INFORMATION ON INGREDIENTS INGREDIENTS: %wt/wt : CAS NUMBER: ACGIH TLV TWA: OSHA PEL TWA OTHER: Tetrahydrofuran 25 – 45% 109-99-9 50 ppm(skin) 200 ppm 25 ppm (Mfg) 100 ppm STEL Methyl Ethyl Ketone 10 – 30% 78-93-3 200 ppm 200 ppm None 300 ppm Acetone 5 - 20% 67-64-1 500 ppm 1000 ppm None 750 ppm STEL CPVC Resin 12 – 20% 68648-82-8 10 mg/m3 None None (Non-hazardous) Established Cyclohexanone 5 – 15% 108-94-1 20 ppm(skin) 50 ppm None 50 ppm STEL Amorphous Fumed Silica 1 - 5% 112945-52-5 10 mg/m3 None None (Non-hazardous) Established a OSHA Hazard Classification: Flammable, irritant, organ effects Section 4 FIRST AID MEASURES Skin: Remove contaminated clothing immediately. -

Peroxide-Forming Chemicals
Peroxide-Forming Chemicals Overview . Only purchase quantities of peroxide-forming chemicals that you expect to use within safe storage timeframes. Peroxide-forming chemicals are a class of compounds that have the ability to form shock-sensitive explosive peroxide crystals. 2. Inventory Requirements Many of the organic solvents commonly used in laboratories at . Confirm that peroxide-forming chemicals are included in Weill Cornell Medicine (WCM) are peroxide formers. the Salute chemical inventory upon receipt, and removed from the inventory when discarded. Typical classes of compounds that form peroxides include: 3. Labeling Requirements . Ethers, acetals, and ketals, especially cyclic ethers and those . All containers of peroxide-forming chemicals must be with primary and/or secondary alkyl groups; labeled with the date received and the date of opening. Aldehydes, including acetaldehyde and benzaldehyde; EHS can provide labels similar to the example below. Compounds containing benzylic hydrogens; and . Compounds containing allylic hydrogens, including most alkenes; vinyl and vinylidene compounds, and dienes. Ethers are the most commonly used peroxide formers at WCM such as tetrahydrofuran, dioxane, diethyl ether, isopropyl ether. Refer to Appendix A for a list of typical peroxide formers found at WCM and guidance on safe storage and testing frequency. PLEASE NOTE: Peroxide crystals may form on the container plug or the threads of the lid, and detonate when the lid is twisted. Do not open a liquid organic peroxide or 4. Storage and Use Requirements peroxide-forming chemical if crystals or a precipitate are . Always consult the manufacturer’s Safety Data Sheet. present. Follow Appendix A safe storage and testing guidelines. Store peroxide formers in original, airtight bottles, away Applicability from light and heat. -

Syntheses of Poly(Ethylene Oxide) Macromonomers Carrying Tertiary Amine and Quaternary Ammonium End Groups
Polymer Journal, Vol.35, No. 6, pp 513—518 (2003) Syntheses of Poly(ethylene oxide) Macromonomers Carrying Tertiary Amine and Quaternary Ammonium End Groups † Takamichi SENYO, Yuji ATAGO, Huanan LIANG, Renhua SHEN, and Koichi ITO Department of Materials Science, Toyohashi University of Technology, Tempaku-cho, Toyohashi 441–8580, Japan (Received January 23, 2003; Accepted March 30, 2003) ABSTRACT: p-Vinylbenzyl alcohol, partially alkoxidated with potassium naphthalene, was used successfully to initiate living polymerization of ethylene oxide to afford α-p-vinylbenzyl-ω-hydroxy poly(ethylene oxide) (PEO) macromonomers. The ω-hydroxy end-groups were quantitatively transformed to tertiary amines either by tosylation followed by reaction with potassium 2-dimethylaminoethoxide or by Williamson synthesis with 2-dimethylaminoethyl chloride in the presence of sodium hydride. ω-Quaternary ammonium-ended PEO macromonomers were also quantita- tively obtained by reaction with iodomethane. KEY WORDS Poly(ethylene oxide) / Macromonomers / p-Vinylbenzyl End-Group / Tertiary Amine End-Group / Quaternary Ammonium End-Group / End-Group Transformation / Hetero- telechelics / Poly(ethylene oxide) (PEO) is one of well- ion to afford F– and –OH end-functionalized PEO, known, water-soluble, nonionic polymers, and its F–O[CH2CH2O]n–H, after acidification. In fact, we macromonomers have also been a subject of consid- could readily have α-p-vinylphenylalkyl-ω-hydroxy- erable interest because of their unique amphiphilic ended PEO macromonomers in one step.13 On the other properties as well as their many potential applica- hand, the ω-hydroxy-end may be transformed to intro- tions in various fields, including coatings, cosmetics, duce another functionality, F , to F–O[CH2CH2O]n–F .