The Heterogeneous Landscape of ALK Negative ALCL
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Applying Expression Profile Similarity for Discovery of Patient-Specific
bioRxiv preprint doi: https://doi.org/10.1101/172015; this version posted September 17, 2017. The copyright holder for this preprint (which was not certified by peer review) is the author/funder, who has granted bioRxiv a license to display the preprint in perpetuity. It is made available under aCC-BY 4.0 International license. Applying expression profile similarity for discovery of patient-specific functional mutations Guofeng Meng Partner Institute of Computational Biology, Yueyang 333, Shanghai, China email: [email protected] Abstract The progress of cancer genome sequencing projects yields unprecedented information of mutations for numerous patients. However, the complexity of mutation profiles of patients hinders the further understanding of mechanisms of oncogenesis. One basic question is how to uncover mutations with functional impacts. In this work, we introduce a computational method to predict functional somatic mutations for each of patient by integrating mutation recurrence with similarity of expression profiles of patients. With this method, the functional mutations are determined by checking the mutation enrichment among a group of patients with similar expression profiles. We applied this method to three cancer types and identified the functional mutations. Comparison of the predictions for three cancer types suggested that most of the functional mutations were cancer-type-specific with one exception to p53. By checking prediction results, we found that our method effectively filtered non-functional mutations resulting from large protein sizes. In addition, this methods can also perform functional annotation to each patient to describe their association with signalling pathways or biological processes. In breast cancer, we predicted "cell adhesion" and other mutated gene associated terms to be significantly enriched among patients. -

Deletions of IKZF1 and SPRED1 Are Associated with Poor Prognosis in A
Leukemia (2014) 28, 302–310 & 2014 Macmillan Publishers Limited All rights reserved 0887-6924/14 www.nature.com/leu ORIGINAL ARTICLE Deletions of IKZF1 and SPRED1 are associated with poor prognosis in a population-based series of pediatric B-cell precursor acute lymphoblastic leukemia diagnosed between 1992 and 2011 L Olsson1, A Castor2, M Behrendtz3, A Biloglav1, E Forestier4, K Paulsson1 and B Johansson1,5 Despite the favorable prognosis of childhood acute lymphoblastic leukemia (ALL), a substantial subset of patients relapses. As this occurs not only in the high risk but also in the standard/intermediate groups, the presently used risk stratification is suboptimal. The underlying mechanisms for treatment failure include the presence of genetic changes causing insensitivity to the therapy administered. To identify relapse-associated aberrations, we performed single-nucleotide polymorphism array analyses of 307 uniformly treated, consecutive pediatric ALL cases accrued during 1992–2011. Recurrent aberrations of 14 genes in patients who subsequently relapsed or had induction failure were detected. Of these, deletions/uniparental isodisomies of ADD3, ATP10A, EBF1, IKZF1, PAN3, RAG1, SPRED1 and TBL1XR1 were significantly more common in B-cell precursor ALL patients who relapsed compared with those remaining in complete remission. In univariate analyses, age (X10 years), white blood cell counts (4100 Â 109/l), t(9;22)(q34;q11), MLL rearrangements, near-haploidy and deletions of ATP10A, IKZF1, SPRED1 and the pseudoautosomal 1 regions on Xp/Yp were significantly associated with decreased 10-year event-free survival, with IKZF1 abnormalities being an independent risk factor in multivariate analysis irrespective of the risk group. Older age and deletions of IKZF1 and SPRED1 were also associated with poor overall survival. -

Comprehensive Genome and Transcriptome Analysis Reveals Genetic Basis for Gene Fusions in Cancer
bioRxiv preprint doi: https://doi.org/10.1101/148684; this version posted June 12, 2017. The copyright holder for this preprint (which was not certified by peer review) is the author/funder, who has granted bioRxiv a license to display the preprint in perpetuity. It is made available under aCC-BY-NC-ND 4.0 International license. Comprehensive genome and transcriptome analysis reveals genetic basis for gene fusions in cancer Nuno A. Fonseca1*, Yao He2*, Liliana Greger1, PCAWG3, Alvis Brazma1, Zemin Zhang2 1 European Molecular Biology Laboratory, European Bioinformatics Institute (EMBL-EBI), Wellcome Trust Genome Campus, Hinxton, Cambridge CB10 1SD, UK; 2 Peking-Tsinghua Centre for Life Sciences, BIOPIC, and Beijing Advanced Innovation Centre for Genomics, Peking University, Beijing, 100871, China *Joint first authors Gene fusions are an important class of cancer-driving events with therapeutic and diagnostic values, yet their underlying genetic mechanisms have not been systematically characterized. Here by combining RNA and whole genome DNA sequencing data from 1188 donors across 27 cancer types we obtained a list of 3297 high-confidence tumour-specific gene fusions, 82% of which had structural variant (SV) support and 2372 of which were novel. Such a large collection of RNA and DNA alterations provides the first opportunity to systematically classify the gene fusions at a mechanistic level. While many could be explained by single SVs, numerous fusions involved series of structural rearrangements and thus are composite fusions. We discovered 75 fusions of a novel class of inter-chromosomal composite fusions, termed bridged fusions, in which a third genomic location bridged two different genes. -

Fusion of the TBL1XR1 and HMGA1 Genes in Splenic Hemangioma with T(3;6)(Q26;P21)
1242 INTERNATIONAL JOURNAL OF ONCOLOGY 48: 1242-1250, 2016 Fusion of the TBL1XR1 and HMGA1 genes in splenic hemangioma with t(3;6)(q26;p21) IOANNIS PANAGOPOULOS1,2, LUDMILA GORUNOVA1,2, BODIL BJERKEHAGEN3, INGVILD LOBMAIER3 and SVERRE HEIM1,2,4 1Section for Cancer Cytogenetics, Institute for Cancer Genetics and Informatics, The Norwegian Radium Hospital, Oslo University Hospital; 2Centre for Cancer Biomedicine, Faculty of Medicine, University of Oslo; 3Department of Pathology, The Norwegian Radium Hospital, Oslo University Hospital; 4Faculty of Medicine, University of Oslo, Oslo, Norway Received September 27, 2015; Accepted November 26, 2015 DOI: 10.3892/ijo.2015.3310 Abstract. RNA-sequencing of a splenic hemangioma with 0.03-14% found in large autopsy series (1,2). In the vast majority the karyotype 45~47,XX,t(3;6)(q26;p21) showed that this of cases, splenic hemangiomas are incidental surgical, radio- translocation generated a chimeric TBL1XR1-HMGA1 gene. logic, or autopsy findings, made during evaluation for other This is the first time that this tumor has been subjected disorders (1-3). Arber et al (3) reported 7 localized splenic to genetic analysis, but the finding of an acquired clonal hemangiomas all of which were discovered incidentally in chromosome abnormality in cells cultured from the lesion surgical patients. In a large series of 32 patients with splenic and the presence of the TBL1XR1-HMGA1 fusion in them hemangioma, only 6 presented with abdominal symptoms and strongly favor the conclusion that splenic hemangiomas are only 4 had a palpable spleen (1). of a neoplastic nature. Genomic PCR confirmed the presence Because of the general absence of presenting symptoms of the TBL1XR1-HMGA1 fusion gene, and RT-PCR together and the typically incidental nature of splenic hemangiomas, the with Sanger sequencing verified the presence of the fusion age at presentation varies considerably. -

Recurrent Mutations of MYD88 and TBL1XR1 in Primary Central Nervous System Lymphomas
Published OnlineFirst July 26, 2012; DOI: 10.1158/1078-0432.CCR-12-0845 Clinical Cancer Human Cancer Biology Research Recurrent Mutations of MYD88 and TBL1XR1 in Primary Central Nervous System Lymphomas Alberto Gonzalez-Aguilar1,2, Ahmed Idbaih1,2, Blandine Boisselier2, Naïma Habbita2, Marta Rossetto2, Alice Laurenge2, Aurelie Bruno2, Anne Jouvet5, Marc Polivka3, Clovis Adam7, Dominique Figarella-Branger8, Catherine Miquel4, Anne Vital9, Herve Ghesquieres 6,Remy Gressin11, Vincent Delwail12, Luc Taillandier13, Olivier Chinot8, Pierre Soubeyran10, Emmanuel Gyan14, Sylvain Choquet1, Caroline Houillier2, Carole Soussain15, Marie-Laure Tanguy2, Yannick Marie2, Karima Mokhtari1,2, and Khe^ Hoang-Xuan1,2 Abstract Purpose: Our objective was to identify the genetic changes involved in primary central nervous system lymphoma (PCNSL) oncogenesis and evaluate their clinical relevance. Experimental Design: We investigated a series of 29 newly diagnosed, HIV-negative, PCNSL patients using high-resolution single-nucleotide polymorphism (SNP) arrays (n ¼ 29) and whole-exome sequencing (n ¼ 4) approaches. Recurrent homozygous deletions and somatic gene mutations found were validated by quantitative real-time PCR and Sanger sequencing, respectively. Molecular results were correlated with prognosis. Results: All PCNSLs were diffuse large B-cell lymphomas, and the patients received chemotherapy without radiotherapy as initial treatment. The SNP analysis revealed recurrent large and focal chromosome imbalances that target candidate genes in PCNSL oncogenesis. The most frequent genomic abnormalities were (i) 6p21.32 loss (HLA locus), (ii) 6q loss, (iii) CDKN2A homozygous deletions, (iv) 12q12-q22, and (v) chromosome 7q21 and 7q31 gains. Homozygous deletions of PRMD1, TOX, and DOCK5 and the amplification of HDAC9 were also detected. Sequencing of matched tumor and blood DNA samples identified novel somatic mutations in MYD88 and TBL1XR1 in 38% and 14% of the cases, respectively. -

Autism and Cancer Share Risk Genes, Pathways, and Drug Targets
TIGS 1255 No. of Pages 8 Forum Table 1 summarizes the characteristics of unclear whether this is related to its signal- Autism and Cancer risk genes for ASD that are also risk genes ing function or a consequence of a second for cancers, extending the original finding independent PTEN activity, but this dual Share Risk Genes, that the PI3K-Akt-mTOR signaling axis function may provide the rationale for the (involving PTEN, FMR1, NF1, TSC1, and dominant role of PTEN in cancer and Pathways, and Drug TSC2) was associated with inherited risk autism. Other genes encoding common Targets for both cancer and ASD [6–9]. Recent tumor signaling pathways include MET8[1_TD$IF],[2_TD$IF] genome-wide exome-sequencing studies PTK7, and HRAS, while p53, AKT, mTOR, Jacqueline N. Crawley,1,2,* of de novo variants in ASD and cancer WNT, NOTCH, and MAPK are compo- Wolf-Dietrich Heyer,3,4 and have begun to uncover considerable addi- nents of signaling pathways regulating Janine M. LaSalle1,4,5 tional overlap. What is surprising about the the nuclear factors described above. genes in Table 1 is not necessarily the Autism is a neurodevelopmental number of risk genes found in both autism Autism is comorbid with several mono- and cancer, but the shared functions of genic neurodevelopmental disorders, disorder, diagnosed behaviorally genes in chromatin remodeling and including Fragile X (FMR1), Rett syndrome by social and communication genome maintenance, transcription fac- (MECP2), Phelan-McDermid (SHANK3), fi de cits, repetitive behaviors, tors, and signal transduction pathways 15q duplication syndrome (UBE3A), and restricted interests. Recent leading to nuclear changes [7,8]. -

Insights Into the Genomic Landscape of MYD88 Wild-Type Waldenström
REGULAR ARTICLE Insights into the genomic landscape of MYD88 wild-type Waldenstrom¨ macroglobulinemia Zachary R. Hunter,1,2 Lian Xu,1 Nickolas Tsakmaklis,1 Maria G. Demos,1 Amanda Kofides,1 Cristina Jimenez,1 Gloria G. Chan,1 Jiaji Chen,1 Xia Liu,1 Manit Munshi,1 Joshua Gustine,1 Kirsten Meid,1 Christopher J. Patterson,1 Guang Yang,1,2 Toni Dubeau,1 Mehmet K. Samur,2,3 Jorge J. Castillo,1,2 Kenneth C. Anderson,2,3 Nikhil C. Munshi,2,3 and Steven P. Treon1,2 1Bing Center for Waldenstrom¨ ’s Macroglobulinemia, Dana-Farber Cancer Institute, Boston, MA; 2Harvard Medical School, Boston, MA; and 3Jerome Lipper Myeloma Center, Dana-Farber Cancer Institute, Boston, MA Activating MYD88 mutations are present in 95% of Waldenstrom¨ macroglobulinemia (WM) Key Points patients, and trigger NF-kB through BTK and IRAK. The BTK inhibitor ibrutinib is active • MUT Mutations affecting in MYD88-mutated (MYD88 ) WM patients, but shows lower activity in MYD88 wild-type k WT WT NF- B, epigenomic (MYD88 ) disease. MYD88 patients also show shorter overall survival, and increased regulation, or DNA risk of disease transformation in some series. The genomic basis for these findings remains damage repair were to be clarified. We performed whole exome and transcriptome sequencing of sorted tumor identified in MYD88 WT samples from 18 MYD88 patients and compared findings with WM patients with wild-type WM. MUT MYD88 disease. We identified somatic mutations predicted to activate NF-kB(TBL1XR1, • k NF- B pathway muta- PTPN13, MALT1, BCL10, NFKB2, NFKBIB, NFKBIZ, and UDRL1F), impart epigenomic tions were downstream dysregulation (KMT2D, KMT2C, and KDM6A), or impair DNA damage repair (TP53, ATM, and of BTK, and many TRRAP). -
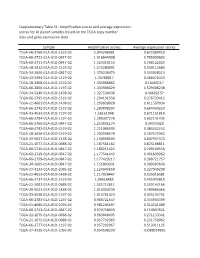
Supplementary Table S1: Amplification Scores and Average Expression Scores for All Paired Samples Based on the TCGA Copy Number Data and Gene Expression Data
Supplementary Table S1: Amplification scores and average expression scores for all paired samples based on the TCGA copy number data and gene expression data Sample Amplification Scores Average expression scores TCGA-46-3768-01A-01D-1519-02 2.045246981 0.672389419 TCGA-60-2722-01A-01D-0847-02 1.916644908 0.785999602 TCGA-60-2711-01A-01D-0847-02 1.523310153 0.766122929 TCGA-18-3412-01A-01D-1519-02 1.472280991 0.349112684 TCGA-34-2600-01A-01D-0847-02 1.370236474 0.343048213 TCGA-43-3394-01A-01D-1519-02 1.34788017 0.466472433 TCGA-18-3408-01A-01D-1519-02 1.334968882 0.16463217 TCGA-66-2800-01A-01D-1197-02 1.333906029 0.529406238 TCGA-34-5240-01A-01D-1438-02 1.327530436 0.48692157 TCGA-66-2795-01A-02D-1519-02 1.294191556 0.276733412 TCGA-22-4601-01A-01D-1438-02 1.293028928 0.611597034 TCGA-66-2742-01A-01D-1519-02 1.283898207 0.684409253 TCGA-33-4533-01A-01D-1197-02 1.26161948 0.671151814 TCGA-66-2794-01A-01D-1197-02 1.260307276 0.602141435 TCGA-66-2766-01A-01D-0847-02 1.253831174 0.39553029 TCGA-66-2792-01A-01D-1519-02 1.211086939 0.484442252 TCGA-18-3416-01A-01D-1519-02 1.204938576 0.267573392 TCGA-39-5037-01A-01D-1438-02 1.190998509 0.807937473 TCGA-21-1077-01A-01D-0688-02 1.187561162 0.825188811 TCGA-60-2710-01A-01D-0847-02 1.180211422 0.209100556 TCGA-60-2719-01A-01D-0847-02 1.177541443 0.491699962 TCGA-66-2759-01A-01D-0847-02 1.177429217 0.368721757 TCGA-34-2605-01A-01D-0847-02 1.151803931 0.309992646 TCGA-37-4133-01A-01D-1095-02 1.124043658 0.227949298 TCGA-22-4613-01A-01D-1438-02 1.117028447 0.535916581 TCGA-66-2737-01A-01D-1519-02 -

Identification of the Global Mir-130A Targetome Reveals a Novel Role for TBL1XR1 in Hematopoietic Stem Cell Self-Renewal and T(8;21) AML
bioRxiv preprint doi: https://doi.org/10.1101/2021.07.30.454489; this version posted July 30, 2021. The copyright holder for this preprint (which was not certified by peer review) is the author/funder. All rights reserved. No reuse allowed without permission. Identification of the Global miR-130a Targetome Reveals a Novel Role for TBL1XR1 in Hematopoietic Stem Cell Self-Renewal and t(8;21) AML Authors: Gabriela Krivdova1,2, Veronique Voisin3, Erwin M Schoof1, Sajid A Marhon1, Alex Murison1, Jessica L McLeod1, Martino Gabra4, Andy GX Zeng1,2, Eric L Van Nostrand5, Stefan Aigner5, Alexander A Shishkin5, Brian A Yee5, Karin G Hermans1,6, Aaron G Trotman-Grant1, Nathan Mbong1, James A Kennedy1,7, Olga I Gan1, Elvin Wagenblast1, Daniel D De Carvalho1,8, Leonardo Salmena1,4, Mark D Minden1, Gary D Bader1,2,3,9, Gene W Yeo5, John E Dick1,2*, Eric R Lechman1*. Affiliations: 1 Princess Margaret Cancer Centre, University Health Network, Toronto, ON M5G 1L7, Canada 2 Department of Molecular Genetics, University of Toronto, Toronto, ON M5S1A5, Canada 3 The Donnelly Centre, University of Toronto, Toronto, ON M5S 3E1, Canada 4 Department of Pharmacology and Toxicology, University of Toronto, Toronto, ON M5S 1A8, Canada 5 Department of Cellular and Molecular Medicine, University of California San Diego, La Jolla, CA 92037, USA 6 Program of Developmental & Stem Cell Biology, Peter Gilgan Centre for Research and Learning, The Hospital for Sick Children, Toronto, ON M5G0A4, Canada 7 Division of Medical Oncology and Hematology, Sunnybrook Health Sciences -

Statistical and Bioinformatic Analysis of Hemimethylation Patterns in Non-Small Cell Lung Cancer Shuying Sun1* , Austin Zane2, Carolyn Fulton3 and Jasmine Philipoom4
Sun et al. BMC Cancer (2021) 21:268 https://doi.org/10.1186/s12885-021-07990-7 RESEARCH ARTICLE Open Access Statistical and bioinformatic analysis of hemimethylation patterns in non-small cell lung cancer Shuying Sun1* , Austin Zane2, Carolyn Fulton3 and Jasmine Philipoom4 Abstract Background: DNA methylation is an epigenetic event involving the addition of a methyl-group to a cytosine- guanine base pair (i.e., CpG site). It is associated with different cancers. Our research focuses on studying non-small cell lung cancer hemimethylation, which refers to methylation occurring on only one of the two DNA strands. Many studies often assume that methylation occurs on both DNA strands at a CpG site. However, recent publications show the existence of hemimethylation and its significant impact. Therefore, it is important to identify cancer hemimethylation patterns. Methods: In this paper, we use the Wilcoxon signed rank test to identify hemimethylated CpG sites based on publicly available non-small cell lung cancer methylation sequencing data. We then identify two types of hemimethylated CpG clusters, regular and polarity clusters, and genes with large numbers of hemimethylated sites. Highly hemimethylated genes are then studied for their biological interactions using available bioinformatics tools. Results: In this paper, we have conducted the first-ever investigation of hemimethylation in lung cancer. Our results show that hemimethylation does exist in lung cells either as singletons or clusters. Most clusters contain only two or three CpG sites. Polarity clusters are much shorter than regular clusters and appear less frequently. The majority of clusters found in tumor samples have no overlap with clusters found in normal samples, and vice versa. -

Characterization of Genomic Alterations in Primary Central Nervous System Lymphomas
Journal of Neuro-Oncology (2018) 140:509–517 https://doi.org/10.1007/s11060-018-2990-6 LABORATORY INVESTIGATION Characterization of genomic alterations in primary central nervous system lymphomas Soheil Zorofchian1 · Hanadi El‑Achi1 · Yuanqing Yan2 · Yoshua Esquenazi2 · Leomar Y. Ballester1,2 Received: 18 June 2018 / Accepted: 22 August 2018 / Published online: 31 August 2018 © Springer Science+Business Media, LLC, part of Springer Nature 2018 Abstract Purpose Primary central nervous system lymphoma (PCNSL) is a non-Hodgkin lymphoma that affects the central nervous system (CNS). Although previous studies have reported the most common mutated genes in PCNSL, including MYD88 and CD79b, our understanding of genetic characterizations in primary CNS lymphomas is limited. The aim of this study was to perform a retrospective analysis investigating the most frequent mutation types, and their frequency, in PCNSL. Methods Fifteen patients with a diagnosis of PCNSL from our institution were analyzed for mutations in 406 genes and rearrangements in 31 genes by next generation sequencing (NGS). Results Missense mutations were identified as the most common mutation type (32%) followed by frame shift mutations (23%). The highest mutation rate was reported in the MYD88 (33.3%), CDKN2A/B (33.3%), and TP53 (26.7%) genes. Inter- mediate tumor mutation burden (TMB) and high TMB was detected in 13.3% and 26.7% of PCNSL, respectively. The most frequent gene rearrangement involved the IGH-BCL6 genes (20%). Conclusions This study shows the most common genetic alterations in PCNSL as determined by a commercial next genera- tion sequencing assay. MYD88 and CD79b are frequently mutated in PCNSL, IGH-BCL6 is the most frequent gene rear- rangement and approximately 1/4 of cases show a high TMB. -

TBL1XR1 As a Potential Therapeutic Target That Promotes Epithelial‑Mesenchymal Transition in Lung Squamous Cell Carcinoma
EXPERIMENTAL AND THERAPEUTIC MEDICINE 17: 91-98, 2019 TBL1XR1 as a potential therapeutic target that promotes epithelial‑mesenchymal transition in lung squamous cell carcinoma YUEHUA ZHAO1,2, HAO LIN1,2, JINGWEI JIANG1,2, MENGXI GE1,2 and XIAOHUA LIANG1,2 1Department of Oncology, Huashan Hospital, Fudan University; 2Department of Oncology, Shanghai Medical College of Fudan University, Shanghai 200040, P.R. China Received May 25, 2018; Accepted August 22, 2018 DOI: 10.3892/etm.2018.6955 Abstract. Transducin (β)-like 1 X-linked receptor 1 Downregulation of TBL1XR1 produced the opposite results. (TBL1XR1) has been demonstrated to serve a vital role in The present study indicated that TBL1XR1 contributed to lung tumor progression. However, the biological role and molecular SCC development and progression, and therefore TBL1XR1 mechanisms of TBL1XR1 in lung squamous cell carcinoma may be a potential therapeutic target. TBL1XR1 may induce (SCC) remain largely unknown. The purpose of the present EMT of lung SCC cells through activation of the TGF-β/Smad study was to investigate the biological role of TBL1XR1 and signaling pathway. its mechanism in lung SCC. TBL1XR1 was expressed in a human bronchial epithelial cell line and in lung SCC cell lines. Introduction The present study analyzed TBL1XR1-induced proliferation, invasion and migration abilities in vitro using the cell counting Lung cancer is the leading cause of cancer mortality and kit-8 assay, cell invasion assay and wound healing assay, is the most frequently diagnosed cancer in the world (1). respectively. This study examined the effects of TBL1XR1 on Annually, there are an estimated 1.8 million new lung cancer epithelial-mesenchymal transition (EMT) in lung SCC cells and cases, accounting for about 13% of total cancer diagnoses (2).