Transcription Factor Activating Protein 4 Is Synthetic Lethal and a Master Regulator of MYCN Amplified Neuroblastoma
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

A Computational Approach for Defining a Signature of Β-Cell Golgi Stress in Diabetes Mellitus
Page 1 of 781 Diabetes A Computational Approach for Defining a Signature of β-Cell Golgi Stress in Diabetes Mellitus Robert N. Bone1,6,7, Olufunmilola Oyebamiji2, Sayali Talware2, Sharmila Selvaraj2, Preethi Krishnan3,6, Farooq Syed1,6,7, Huanmei Wu2, Carmella Evans-Molina 1,3,4,5,6,7,8* Departments of 1Pediatrics, 3Medicine, 4Anatomy, Cell Biology & Physiology, 5Biochemistry & Molecular Biology, the 6Center for Diabetes & Metabolic Diseases, and the 7Herman B. Wells Center for Pediatric Research, Indiana University School of Medicine, Indianapolis, IN 46202; 2Department of BioHealth Informatics, Indiana University-Purdue University Indianapolis, Indianapolis, IN, 46202; 8Roudebush VA Medical Center, Indianapolis, IN 46202. *Corresponding Author(s): Carmella Evans-Molina, MD, PhD ([email protected]) Indiana University School of Medicine, 635 Barnhill Drive, MS 2031A, Indianapolis, IN 46202, Telephone: (317) 274-4145, Fax (317) 274-4107 Running Title: Golgi Stress Response in Diabetes Word Count: 4358 Number of Figures: 6 Keywords: Golgi apparatus stress, Islets, β cell, Type 1 diabetes, Type 2 diabetes 1 Diabetes Publish Ahead of Print, published online August 20, 2020 Diabetes Page 2 of 781 ABSTRACT The Golgi apparatus (GA) is an important site of insulin processing and granule maturation, but whether GA organelle dysfunction and GA stress are present in the diabetic β-cell has not been tested. We utilized an informatics-based approach to develop a transcriptional signature of β-cell GA stress using existing RNA sequencing and microarray datasets generated using human islets from donors with diabetes and islets where type 1(T1D) and type 2 diabetes (T2D) had been modeled ex vivo. To narrow our results to GA-specific genes, we applied a filter set of 1,030 genes accepted as GA associated. -

4-6 Weeks Old Female C57BL/6 Mice Obtained from Jackson Labs Were Used for Cell Isolation
Methods Mice: 4-6 weeks old female C57BL/6 mice obtained from Jackson labs were used for cell isolation. Female Foxp3-IRES-GFP reporter mice (1), backcrossed to B6/C57 background for 10 generations, were used for the isolation of naïve CD4 and naïve CD8 cells for the RNAseq experiments. The mice were housed in pathogen-free animal facility in the La Jolla Institute for Allergy and Immunology and were used according to protocols approved by the Institutional Animal Care and use Committee. Preparation of cells: Subsets of thymocytes were isolated by cell sorting as previously described (2), after cell surface staining using CD4 (GK1.5), CD8 (53-6.7), CD3ε (145- 2C11), CD24 (M1/69) (all from Biolegend). DP cells: CD4+CD8 int/hi; CD4 SP cells: CD4CD3 hi, CD24 int/lo; CD8 SP cells: CD8 int/hi CD4 CD3 hi, CD24 int/lo (Fig S2). Peripheral subsets were isolated after pooling spleen and lymph nodes. T cells were enriched by negative isolation using Dynabeads (Dynabeads untouched mouse T cells, 11413D, Invitrogen). After surface staining for CD4 (GK1.5), CD8 (53-6.7), CD62L (MEL-14), CD25 (PC61) and CD44 (IM7), naïve CD4+CD62L hiCD25-CD44lo and naïve CD8+CD62L hiCD25-CD44lo were obtained by sorting (BD FACS Aria). Additionally, for the RNAseq experiments, CD4 and CD8 naïve cells were isolated by sorting T cells from the Foxp3- IRES-GFP mice: CD4+CD62LhiCD25–CD44lo GFP(FOXP3)– and CD8+CD62LhiCD25– CD44lo GFP(FOXP3)– (antibodies were from Biolegend). In some cases, naïve CD4 cells were cultured in vitro under Th1 or Th2 polarizing conditions (3, 4). -

Genome-Scale Identification of Transcription Factors That Mediate An
ARTICLE DOI: 10.1038/s41467-018-04406-2 OPEN Genome-scale identification of transcription factors that mediate an inflammatory network during breast cellular transformation Zhe Ji 1,2,4, Lizhi He1, Asaf Rotem1,2,5, Andreas Janzer1,6, Christine S. Cheng2,7, Aviv Regev2,3 & Kevin Struhl 1 Transient activation of Src oncoprotein in non-transformed, breast epithelial cells can initiate an epigenetic switch to the stably transformed state via a positive feedback loop that involves 1234567890():,; the inflammatory transcription factors STAT3 and NF-κB. Here, we develop an experimental and computational pipeline that includes 1) a Bayesian network model (AccessTF) that accurately predicts protein-bound DNA sequence motifs based on chromatin accessibility, and 2) a scoring system (TFScore) that rank-orders transcription factors as candidates for being important for a biological process. Genetic experiments validate TFScore and suggest that more than 40 transcription factors contribute to the oncogenic state in this model. Interestingly, individual depletion of several of these factors results in similar transcriptional profiles, indicating that a complex and interconnected transcriptional network promotes a stable oncogenic state. The combined experimental and computational pipeline represents a general approach to comprehensively identify transcriptional regulators important for a biological process. 1 Department of Biological Chemistry and Molecular Pharmacology, Harvard Medical School, Boston, MA 02115, USA. 2 Broad Institute of MIT and Harvard, Cambridge, MA 02142, USA. 3 Department of Biology, Howard Hughes Medical Institute and David H. Koch Institute for Integrative Cancer Research, Massachusetts Institute of Technology, Cambridge, MA 20140, USA. 4Present address: Department of Pharmacology and Biomedical Engineering, Northwestern University, Evanston 60611 IL, USA. -

Quantigene Flowrna Probe Sets Currently Available
QuantiGene FlowRNA Probe Sets Currently Available Accession No. Species Symbol Gene Name Catalog No. NM_003452 Human ZNF189 zinc finger protein 189 VA1-10009 NM_000057 Human BLM Bloom syndrome VA1-10010 NM_005269 Human GLI glioma-associated oncogene homolog (zinc finger protein) VA1-10011 NM_002614 Human PDZK1 PDZ domain containing 1 VA1-10015 NM_003225 Human TFF1 Trefoil factor 1 (breast cancer, estrogen-inducible sequence expressed in) VA1-10016 NM_002276 Human KRT19 keratin 19 VA1-10022 NM_002659 Human PLAUR plasminogen activator, urokinase receptor VA1-10025 NM_017669 Human ERCC6L excision repair cross-complementing rodent repair deficiency, complementation group 6-like VA1-10029 NM_017699 Human SIDT1 SID1 transmembrane family, member 1 VA1-10032 NM_000077 Human CDKN2A cyclin-dependent kinase inhibitor 2A (melanoma, p16, inhibits CDK4) VA1-10040 NM_003150 Human STAT3 signal transducer and activator of transcripton 3 (acute-phase response factor) VA1-10046 NM_004707 Human ATG12 ATG12 autophagy related 12 homolog (S. cerevisiae) VA1-10047 NM_000737 Human CGB chorionic gonadotropin, beta polypeptide VA1-10048 NM_001017420 Human ESCO2 establishment of cohesion 1 homolog 2 (S. cerevisiae) VA1-10050 NM_197978 Human HEMGN hemogen VA1-10051 NM_001738 Human CA1 Carbonic anhydrase I VA1-10052 NM_000184 Human HBG2 Hemoglobin, gamma G VA1-10053 NM_005330 Human HBE1 Hemoglobin, epsilon 1 VA1-10054 NR_003367 Human PVT1 Pvt1 oncogene homolog (mouse) VA1-10061 NM_000454 Human SOD1 Superoxide dismutase 1, soluble (amyotrophic lateral sclerosis 1 (adult)) -
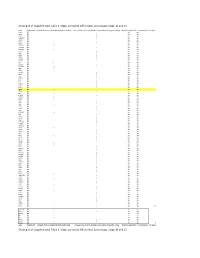
Supplemental Table 3. Stage-Persistent Differential Exon Usage, Stage 10 and 15
DeJong et al. Supplemental Table 3. Stage-persistent Differential Exon Usage, Stage 10 and 15 Gene Condition UP Retained intron as visualized by DEXseq gene modeling Increased levels of intron detected as visualized by DEXseq gene modeling Maternally deposited? > 2 Fold increase? # of genes actr3 MO x yes yes aldoa MO x yes yes alms1 MO x yes yes arhgap12 MO x yes yes arrdc2 MO x yes yes asxl1 MO x yes yes bmp7.1 MO x yes yes c11orf2 MO x yes yes c16orf72 MO x yes yes c1orf112 MO x yes yes caprin1 MO x yes yes cdc6 MO x yes yes cdk12 MO x yes yes cecr2 MO x yes yes cep135 MO x yes yes chmp5 MO x yes yes clp1 MO x yes yes ctdsp2 MO x yes yes dennd2a MO x yes yes dgka MO x yes yes dido1 MO x yes yes dnajc5g MO x yes yes dus4l MO x yes yes eed MO x yes yes eif2s2 MO x yes yes ets1 MO x yes yes ets2 MO x yes yes exosc2 MO x yes yes extl3 MO x yes yes farsa MO x yes yes fgfr4 MO x yes yes fmnl3 MO x yes yes gps2 MO x yes yes gtf2h4 MO x yes yes herpud1 MO x yes yes hexdc MO x yes yes hras MO x yes yes ifrd2 MO x yes yes ing4 MO x yes yes ints6 MO x yes yes irf6.2 MO x yes yes jag1 MO x yes yes kiaa0664 MO x yes yes klf2 MO x yes yes klhdc3 MO x yes yes lemd3 MO x yes yes lrig3 MO x yes yes mcm3ap MO x yes yes myd88 MO x yes yes myo1e.2 MO x yes yes neo1 MO x yes yes nme2 MO x yes yes npdc1.1 MO x yes yes nxf1 MO x yes yes pan3 MO x yes yes parp1 MO x yes yes pask MO x yes yes paxip1 MO x yes yes pdgfa MO x yes yes per2 MO x yes yes per3 MO x yes yes pggt1b MO x yes yes phf8 MO x yes yes pip5k1a MO x yes yes plekhg5 MO x yes yes poldip3 MO x -

The Kinesin Spindle Protein Inhibitor Filanesib Enhances the Activity of Pomalidomide and Dexamethasone in Multiple Myeloma
Plasma Cell Disorders SUPPLEMENTARY APPENDIX The kinesin spindle protein inhibitor filanesib enhances the activity of pomalidomide and dexamethasone in multiple myeloma Susana Hernández-García, 1 Laura San-Segundo, 1 Lorena González-Méndez, 1 Luis A. Corchete, 1 Irena Misiewicz- Krzeminska, 1,2 Montserrat Martín-Sánchez, 1 Ana-Alicia López-Iglesias, 1 Esperanza Macarena Algarín, 1 Pedro Mogollón, 1 Andrea Díaz-Tejedor, 1 Teresa Paíno, 1 Brian Tunquist, 3 María-Victoria Mateos, 1 Norma C Gutiérrez, 1 Elena Díaz- Rodriguez, 1 Mercedes Garayoa 1* and Enrique M Ocio 1* 1Centro Investigación del Cáncer-IBMCC (CSIC-USAL) and Hospital Universitario-IBSAL, Salamanca, Spain; 2National Medicines Insti - tute, Warsaw, Poland and 3Array BioPharma, Boulder, Colorado, USA *MG and EMO contributed equally to this work ©2017 Ferrata Storti Foundation. This is an open-access paper. doi:10.3324/haematol. 2017.168666 Received: March 13, 2017. Accepted: August 29, 2017. Pre-published: August 31, 2017. Correspondence: [email protected] MATERIAL AND METHODS Reagents and drugs. Filanesib (F) was provided by Array BioPharma Inc. (Boulder, CO, USA). Thalidomide (T), lenalidomide (L) and pomalidomide (P) were purchased from Selleckchem (Houston, TX, USA), dexamethasone (D) from Sigma-Aldrich (St Louis, MO, USA) and bortezomib from LC Laboratories (Woburn, MA, USA). Generic chemicals were acquired from Sigma Chemical Co., Roche Biochemicals (Mannheim, Germany), Merck & Co., Inc. (Darmstadt, Germany). MM cell lines, patient samples and cultures. Origin, authentication and in vitro growth conditions of human MM cell lines have already been characterized (17, 18). The study of drug activity in the presence of IL-6, IGF-1 or in co-culture with primary bone marrow mesenchymal stromal cells (BMSCs) or the human mesenchymal stromal cell line (hMSC–TERT) was performed as described previously (19, 20). -

Homologs of Genes Expressed in Caenorhabditis Elegans Gabaergic
Hammock et al. Neural Development 2010, 5:32 http://www.neuraldevelopment.com/content/5/1/32 RESEARCH ARTICLE Open Access Homologs of genes expressed in Caenorhabditis elegans GABAergic neurons are also found in the developing mouse forebrain Elizabeth AD Hammock1,2*, Kathie L Eagleson3, Susan Barlow4,6, Laurie R Earls4,7, David M Miller III2,4,5, Pat Levitt3* Abstract Background: In an effort to identify genes that specify the mammalian forebrain, we used a comparative approach to identify mouse homologs of transcription factors expressed in developing Caenorhabditis elegans GABAergic neurons. A cell-specific microarray profiling study revealed a set of transcription factors that are highly expressed in embryonic C. elegans GABAergic neurons. Results: Bioinformatic analyses identified mouse protein homologs of these selected transcripts and their expression pattern was mapped in the mouse embryonic forebrain by in situ hybridization. A review of human homologs indicates several of these genes are potential candidates in neurodevelopmental disorders. Conclusions: Our comparative approach has revealed several novel candidates that may serve as future targets for studies of mammalian forebrain development. Background As with other cell types, the diversity of GABAergic Proper forebrain patterning and cell-fate specification neurons has its basis in different developmental origins, lay the foundation for complex behaviors. These neuro- with timing and location of birth playing key roles in developmental events in large part depend on a series of cell fate [1,6-8]. gene expression refinements (reviewed in [1]) that com- Despite the phenotypic variety of GABAergic neurons, mit cells to express certain phenotypic features that all use GABA as a neurotransmitter. -

(B6;129.Cg-Gt(ROSA)26Sor Tm20(CAG-Ctgf-GFP)Jsd) Were Crossed with Female Foxd1cre/+ Heterozygote Mice 1, and Experimental Mice Were Selected As Foxd1cre/+; Rs26cig/+
Supplemental Information SI Methods Animal studies Heterozygote mice (B6;129.Cg-Gt(ROSA)26Sor tm20(CAG-Ctgf-GFP)Jsd) were crossed with female Foxd1Cre/+ heterozygote mice 1, and experimental mice were selected as Foxd1Cre/+; Rs26CIG/+. In some studies Coll-GFPTg or TCF/Lef:H2B-GFPTg mice or Foxd1Cre/+; Rs26tdTomatoR/+ mice were used as described 2; 3. Left kidneys were subjected to ureteral obstruction using a posterior surgical approach as described 2. In some experiments recombinant mouse DKK1 (0.5mg/kg) or an equal volume of vehicle was administered by daily IP injection. In the in vivo ASO experiment, either specific Lrp6 (TACCTCAATGCGATTT) or scrambled negative control ASO (AACACGTCTATACGC) (30mg/kg) (Exiqon, LNA gapmers) was administered by IP injection on d-1, d1, d4, and d7. In other experiments anti-CTGF domain-IV antibodies (5mg/kg) or control IgG were administered d-1, d1 and d6. All animal experiments were performed under approved IACUC protocols held at the University of Washington and Biogen. Recombinant protein and antibody generation and characterization Human CTGF domain I (sequence Met1 CPDEPAPRCPAGVSLVLDGCGCCRVCAKQLGELCTERDPCDPHKGLFC), domain I+II (sequence Met1CPDEPAPRCPAGVSLVLDGCGCCRVCAKQLGELCTERDPCDPHKGLFCCIFGGT VYRSGESFQSSCKYQCTCLDGAVGCMPLCSMDVRLPSPDCPFPRRVKLPGKCCEE) were cloned and expressed in 293 cells, and purified by Chelating SFF(Ni) Column, tested for single band by SEC and PAGE, and tested for absence of contamination. Domain-IV (sequence GKKCIRTPKISKPIKFELSGCTSMKTYRAKFCGVCTDGRCCTPHRTTTLPVEFKCPDGE VMKKNMMFIKTCACHYNCPGDNDIFESLYYRKMY) was purchased from Peprotech. Mouse or human DKK1 was generated from the coding sequence with some modifications and a tag. Secreted protein was harvested from 293 cells, and purified by nickel column, and tested for activity in a supertopflash (STF) assay 4. DKK1 showed EC50 of 0.69nM for WNT3a-induced WNT signaling in STF cells. -

Positional Specificity of Different Transcription Factor Classes Within Enhancers
Positional specificity of different transcription factor classes within enhancers Sharon R. Grossmana,b,c, Jesse Engreitza, John P. Raya, Tung H. Nguyena, Nir Hacohena,d, and Eric S. Landera,b,e,1 aBroad Institute of MIT and Harvard, Cambridge, MA 02142; bDepartment of Biology, Massachusetts Institute of Technology, Cambridge, MA 02139; cProgram in Health Sciences and Technology, Harvard Medical School, Boston, MA 02215; dCancer Research, Massachusetts General Hospital, Boston, MA 02114; and eDepartment of Systems Biology, Harvard Medical School, Boston, MA 02215 Contributed by Eric S. Lander, June 19, 2018 (sent for review March 26, 2018; reviewed by Gioacchino Natoli and Alexander Stark) Gene expression is controlled by sequence-specific transcription type-restricted enhancers (active in <50% of the cell types) and factors (TFs), which bind to regulatory sequences in DNA. TF ubiquitous enhancers (active in >90% of the cell types) (SI Ap- binding occurs in nucleosome-depleted regions of DNA (NDRs), pendix, Fig. S1C). which generally encompass regions with lengths similar to those We next sought to infer functional TF-binding sites within the protected by nucleosomes. However, less is known about where active regulatory elements. In a recent study (5), we found that within these regions specific TFs tend to be found. Here, we char- TF binding is strongly correlated with the quantitative DNA acterize the positional bias of inferred binding sites for 103 TFs accessibility of a region. Furthermore, the TF motifs associated within ∼500,000 NDRs across 47 cell types. We find that distinct with enhancer activity in reporter assays in a cell type corre- classes of TFs display different binding preferences: Some tend to sponded closely to those that are most enriched in the genomic have binding sites toward the edges, some toward the center, and sequences of active regulatory elements in that cell type (5). -

Regulatory Network Analysis of Genes and Micrornas in Human Hepatoblastoma
ONCOLOGY LETTERS 12: 4099-4106, 2016 Regulatory network analysis of genes and microRNAs in human hepatoblastoma JIMIN HE1,3, XIAOXIN GUO1-3, LINLIN SUN2,3, NING WANG1,3 and JIWEI BAO2,3 1College of Computer Science and Technology; 2College of Software; 3Key Laboratory of Symbolic Computation and Knowledge Engineering of Ministry of Education, Jilin University, Changchun, Jilin 130012, P.R. China Received March 12, 2015; Accepted January 11, 2016 DOI: 10.3892/ol.2016.5196 Abstract. Hepatoblastoma (HB) is a common type of primary Homo sapiens (hsa)-miR-221, hsa-miR-18a and hsa-miR-17-5p, tumor in children. Previous studies have examined the expression but no miRNAs targeted MYCN. In conclusion, the pathways of genes, including transcription factors (TFs), target genes, host and mechanisms underlying HB were expounded in the present genes and microRNAs (miRNAs or miRs) associated with HB. study, which proposed a fundamental hypothesis for additional However, the regulatory pathways of miRNAs and genes remain studies. unclear. In the present study, a novel perspective is proposed, which focuses on HB and the associated regulatory pathways, Introduction to construct three networks at various levels, including a differ- entially expressed network, an associated network and a global Hepatoblastoma (HB) is the most common hepatic malig- network. Genes and miRNAs are considered as key factors in the nancy in children, and accounts for ~1% of all childhood network. In the three networks, the associations between each tumors (1). Usually, the disease is diagnosed during the first pair of factors, including TFs that regulate miRNAs, miRNAs three years of a child's life (2). -

(12) Patent Application Publication (10) Pub. No.: US 2009/0269772 A1 Califano Et Al
US 20090269772A1 (19) United States (12) Patent Application Publication (10) Pub. No.: US 2009/0269772 A1 Califano et al. (43) Pub. Date: Oct. 29, 2009 (54) SYSTEMS AND METHODS FOR Publication Classification IDENTIFYING COMBINATIONS OF (51) Int. Cl. COMPOUNDS OF THERAPEUTIC INTEREST CI2O I/68 (2006.01) CI2O 1/02 (2006.01) (76) Inventors: Andrea Califano, New York, NY G06N 5/02 (2006.01) (US); Riccardo Dalla-Favera, New (52) U.S. Cl. ........... 435/6: 435/29: 706/54; 707/E17.014 York, NY (US); Owen A. (57) ABSTRACT O'Connor, New York, NY (US) Systems, methods, and apparatus for searching for a combi nation of compounds of therapeutic interest are provided. Correspondence Address: Cell-based assays are performed, each cell-based assay JONES DAY exposing a different sample of cells to a different compound 222 EAST 41ST ST in a plurality of compounds. From the cell-based assays, a NEW YORK, NY 10017 (US) Subset of the tested compounds is selected. For each respec tive compound in the Subset, a molecular abundance profile from cells exposed to the respective compound is measured. (21) Appl. No.: 12/432,579 Targets of transcription factors and post-translational modu lators of transcription factor activity are inferred from the (22) Filed: Apr. 29, 2009 molecular abundance profile data using information theoretic measures. This data is used to construct an interaction net Related U.S. Application Data work. Variances in edges in the interaction network are used to determine the drug activity profile of compounds in the (60) Provisional application No. 61/048.875, filed on Apr. -

Autocrine IFN Signaling Inducing Profibrotic Fibroblast Responses By
Downloaded from http://www.jimmunol.org/ by guest on September 23, 2021 Inducing is online at: average * The Journal of Immunology , 11 of which you can access for free at: 2013; 191:2956-2966; Prepublished online 16 from submission to initial decision 4 weeks from acceptance to publication August 2013; doi: 10.4049/jimmunol.1300376 http://www.jimmunol.org/content/191/6/2956 A Synthetic TLR3 Ligand Mitigates Profibrotic Fibroblast Responses by Autocrine IFN Signaling Feng Fang, Kohtaro Ooka, Xiaoyong Sun, Ruchi Shah, Swati Bhattacharyya, Jun Wei and John Varga J Immunol cites 49 articles Submit online. Every submission reviewed by practicing scientists ? is published twice each month by Receive free email-alerts when new articles cite this article. Sign up at: http://jimmunol.org/alerts http://jimmunol.org/subscription Submit copyright permission requests at: http://www.aai.org/About/Publications/JI/copyright.html http://www.jimmunol.org/content/suppl/2013/08/20/jimmunol.130037 6.DC1 This article http://www.jimmunol.org/content/191/6/2956.full#ref-list-1 Information about subscribing to The JI No Triage! Fast Publication! Rapid Reviews! 30 days* Why • • • Material References Permissions Email Alerts Subscription Supplementary The Journal of Immunology The American Association of Immunologists, Inc., 1451 Rockville Pike, Suite 650, Rockville, MD 20852 Copyright © 2013 by The American Association of Immunologists, Inc. All rights reserved. Print ISSN: 0022-1767 Online ISSN: 1550-6606. This information is current as of September 23, 2021. The Journal of Immunology A Synthetic TLR3 Ligand Mitigates Profibrotic Fibroblast Responses by Inducing Autocrine IFN Signaling Feng Fang,* Kohtaro Ooka,* Xiaoyong Sun,† Ruchi Shah,* Swati Bhattacharyya,* Jun Wei,* and John Varga* Activation of TLR3 by exogenous microbial ligands or endogenous injury-associated ligands leads to production of type I IFN.