A Molecular Phylogeny of the Pheasants and Partridges Suggests That These Lineages Are Not Monophyletic R
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-
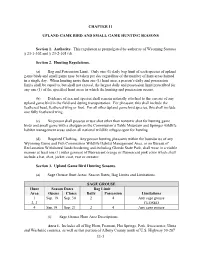
Sage-Grouse Hunting Season
CHAPTER 11 UPLAND GAME BIRD AND SMALL GAME HUNTING SEASONS Section 1. Authority. This regulation is promulgated by authority of Wyoming Statutes § 23-1-302 and § 23-2-105 (d). Section 2. Hunting Regulations. (a) Bag and Possession Limit. Only one (1) daily bag limit of each species of upland game birds and small game may be taken per day regardless of the number of hunt areas hunted in a single day. When hunting more than one (1) hunt area, a person’s daily and possession limits shall be equal to, but shall not exceed, the largest daily and possession limit prescribed for any one (1) of the specified hunt areas in which the hunting and possession occurs. (b) Evidence of sex and species shall remain naturally attached to the carcass of any upland game bird in the field and during transportation. For pheasant, this shall include the feathered head, feathered wing or foot. For all other upland game bird species, this shall include one fully feathered wing. (c) No person shall possess or use shot other than nontoxic shot for hunting game birds and small game with a shotgun on the Commission’s Table Mountain and Springer wildlife habitat management areas and on all national wildlife refuges open for hunting. (d) Required Clothing. Any person hunting pheasants within the boundaries of any Wyoming Game and Fish Commission Wildlife Habitat Management Area, or on Bureau of Reclamation Withdrawal lands bordering and including Glendo State Park, shall wear in a visible manner at least one (1) outer garment of fluorescent orange or fluorescent pink color which shall include a hat, shirt, jacket, coat, vest or sweater. -

Assessing the Status of Handsome Francolin Francolinus Nobilis in Bwindi Impenetrable National Park, Western Uganda
Scopus 25: 4150, December 2005 Assessing the status of Handsome Francolin Francolinus nobilis in Bwindi Impenetrable National Park, western Uganda Richard Ssemmanda and Richard A. Fuller The Handsome Francolin is a ground-dwelling partridge occurring in montane forest and the high-altitude bamboo zone within a restricted area along the Albertine Rift Mountains from the Bleus Mountains in eastern Democratic Republic of the Congo (DRC) south to Burundi (Urban et al. 1986, Madge & McGowan 2002). It appears to occur over a global range of about 120 000 km2 (Fuller et al. 2000, BirdLife International 2004), although there are very few recent records from most parts of this area. The small area of the Albertine Rift Mountains supports 41 endemic bird species, 11 of which are globally threatened (BirdLife International 2004, Plumptre et al. 2003). Forest covers much of the 56 000 km2 in the Albertine Rift Endemic Bird Area, and much of it is rugged terrain very difficult to access (Shaw & Shewry 2001). Despite this, there are significant human pressures in the area, stemming mostly from large increases in human population densities as a result of refugee movements (Omari et al. 1999, Gatarabirwa et al. 2000). Being large, slow-moving, palatable terrestrial birds, the Galliformes are under particularly direct pressure from humans through hunting and disturbance, and they may provide useful rapid indicators of the amount of human pressure in a particular area. The area surrounding Bwindi is one of Ugandas most densely populated rural areas with human densities of 160320 people/km2. Approximately 10 000 families cultivate the land immediately surrounding the park (Butynski 1984). -

Congo Peacock – an Update
Congo Peacock – an update The discovery of Congo Peacock Afropavo congensis was one of the most sensational ornithological events of the 20 th century, partly because such a large bird had eluded discovery and partly because of its potential affinity with Asiatic species (1). It was in 1936 that James Chapin found two mounted specimens put away on top of a cabinet at the Belgian Congo Museum (now Royal Museum for Central Africa in Tervuren, near Brussels), having been misidentified as Pavo cristatus for 20 years, and thus considered uninteresting (2). It was rediscovered and photographed by wildlife film-maker Alan Root in 1993 (3, 4). It has remained an elusive and rarely seen and photographed species so it was with great interest and excitement to receive this photograph of a female Congo Peacock located by Mark Van Beirs (http://www.birdquest-tours.com/news ) ABC member Michel Louette, the former head of Ornithology at the RMCA writes to say that his late student Emile Mulotwa at the University of Kisangani, worked on this species for his doctoral thesis in 2008. During this work, he made many sightings (5-7). But Emile died sadly in a plane accident in 2011. References 1. Urban, E.K., Fry, C.H. and Keith, S. (1986) The Birds of Africa Volume II, pp. 11-13. 2. Chapin, J.P. (1936) A new peacock-like bird from the Belgian Congo. Rev. Zool. Bot. Afr. 29, pp. 1-6. 3. Fanshawe, J.H. (1994): Elusive Congo Peacock found by filmmaker Alan Root. ABC Bulletin 1(1) p 27. -

Wild Turkey Education Guide
Table of Contents Section 1: Eastern Wild Turkey Ecology 1. Eastern Wild Turkey Quick Facts………………………………………………...pg 2 2. Eastern Wild Turkey Fact Sheet………………………………………………….pg 4 3. Wild Turkey Lifecycle……………………………………………………………..pg 8 4. Eastern Wild Turkey Adaptations ………………………………………………pg 9 Section 2: Eastern Wild Turkey Management 1. Wild Turkey Management Timeline…………………….……………………….pg 18 2. History of Wild Turkey Management …………………...…..…………………..pg 19 3. Modern Wild Turkey Management in Maryland………...……………………..pg 22 4. Managing Wild Turkeys Today ……………………………………………….....pg 25 Section 3: Activity Lesson Plans 1. Activity: Growing Up WILD: Tasty Turkeys (Grades K-2)……………..….…..pg 33 2. Activity: Calling All Turkeys (Grades K-5)………………………………..…….pg 37 3. Activity: Fit for a Turkey (Grades 3-5)…………………………………………...pg 40 4. Activity: Project WILD adaptation: Too Many Turkeys (Grades K-5)…..…….pg 43 5. Activity: Project WILD: Quick, Frozen Critters (Grades 5-8).……………….…pg 47 6. Activity: Project WILD: Turkey Trouble (Grades 9-12………………….……....pg 51 7. Activity: Project WILD: Let’s Talk Turkey (Grades 9-12)..……………..………pg 58 Section 4: Additional Activities: 1. Wild Turkey Ecology Word Find………………………………………….…….pg 66 2. Wild Turkey Management Word Find………………………………………….pg 68 3. Turkey Coloring Sheet ..………………………………………………………….pg 70 4. Turkey Coloring Sheet ..………………………………………………………….pg 71 5. Turkey Color-by-Letter……………………………………..…………………….pg 72 6. Five Little Turkeys Song Sheet……. ………………………………………….…pg 73 7. Thankful Turkey…………………..…………………………………………….....pg 74 8. Graph-a-Turkey………………………………….…………………………….…..pg 75 9. Turkey Trouble Maze…………………………………………………………..….pg 76 10. What Animals Made These Tracks………………………………………….……pg 78 11. Drinking Straw Turkey Call Craft……………………………………….….……pg 80 Section 5: Wild Turkey PowerPoint Slide Notes The facilities and services of the Maryland Department of Natural Resources are available to all without regard to race, color, religion, sex, sexual orientation, age, national origin or physical or mental disability. -

Than a Meal: the Turkey in History, Myth
More Than a Meal Abigail at United Poultry Concerns’ Thanksgiving Party Saturday, November 22, 1997. Photo: Barbara Davidson, The Washington Times, 11/27/97 More Than a Meal The Turkey in History, Myth, Ritual, and Reality Karen Davis, Ph.D. Lantern Books New York A Division of Booklight Inc. Lantern Books One Union Square West, Suite 201 New York, NY 10003 Copyright © Karen Davis, Ph.D. 2001 All rights reserved. No part of this book may be reproduced, stored in a retrieval system, or transmitted in any form or by any means, electronic, mechanical, photocopying, recording, or otherwise, without the written permission of Lantern Books. Printed in the United States of America Library of Congress Cataloging-in-Publication Data For Boris, who “almost got to be The real turkey inside of me.” From Boris, by Terry Kleeman and Marie Gleason Anne Shirley, 16-year-old star of “Anne of Green Gables” (RKO-Radio) on Thanksgiving Day, 1934 Photo: Underwood & Underwood, © 1988 Underwood Photo Archives, Ltd., San Francisco Table of Contents 1 Acknowledgments . .9 Introduction: Milton, Doris, and Some “Turkeys” in Recent American History . .11 1. A History of Image Problems: The Turkey as a Mock Figure of Speech and Symbol of Failure . .17 2. The Turkey By Many Other Names: Confusing Nomenclature and Species Identification Surrounding the Native American Bird . .25 3. A True Original Native of America . .33 4. Our Token of Festive Joy . .51 5. Why Do We Hate This Celebrated Bird? . .73 6. Rituals of Spectacular Humiliation: An Attempt to Make a Pathetic Situation Seem Funny . .99 7 8 More Than a Meal 7. -

Recording Some of Breeding Birds in Mehmedan Region of Republic Yemen
Available online a t www.pelagiaresearchlibrary.com Pelagia Research Library European Journal of Experimental Biology, 2014, 4(1):625-632 ISSN: 2248 –9215 CODEN (USA): EJEBAU Recording some of breeding birds in Mehmedan region of Republic Yemen Fadhl Adullah Nasser Balem and Mohamed Saleh Alzokary Biology Department, Aden University, Yaman _____________________________________________________________________________________________ ABSTRACT Mehmedan region is always green and there are different trees, shrubs, herbs and a lot of land which cultivated by corn, millet and other monetary plants. The site has been identified by the authors as an important Bird Area and especially for passerines breeding birds. Aim of this paper is to recording of some breeding birds.Many field visits during the year (2012) were conducted and (13) breeding bird species were recoded, these birds relating to (5) Orders, (10) Families, and (11) Genera. Key words: Breeding birds, Mehmedan, Yemen. _____________________________________________________________________________________________ INTRODUCTION At present time about (432) bird species were recorded in avifauna of Yemen of which (1) is endemic, (2) have been introduced by humans, and (25) are rare or accidental, (14) species are globally threatened.Mehmedan region located in southern Tehama which defined as lying south of (21 0N) along the Saudi Arabian and Yemen Red Sea lowlands and east along the Gulf of Aden to approximately (46 0E).Temperatures and humidity greatly increase southwards and rainfall decreases but the area has many permanent water courses and much subsurface water due to the considerable rub-off of rainwater from the highlands. Consequently there is much more vegetation in the wadis and there is a good deal of traditional, small scale agriculture mostly of millet, sorghum and vegetables[1]. -

Reproductive Ecology of Tibetan Eared Pheasant Crossoptilon Harmani in Scrub Environment, with Special Reference to the Effect of Food
Ibis (2003), 145, 657–666 Blackwell Publishing Ltd. Reproductive ecology of Tibetan Eared Pheasant Crossoptilon harmani in scrub environment, with special reference to the effect of food XIN LU1* & GUANG-MEI ZHENG2 1Department of Zoology, College of Life Sciences, Wuhan University, Wuhan 430072, China 2Department of Ecology, College of Life Sciences, Beijing Normal University, Beijing 100875, China We studied the nesting ecology of two groups of the endangered Tibetan Eared Pheasants Crossoptilon harmani in scrub environments near Lhasa, Tibet, during 1996 and 1999–2001. One group received artificial food from a nunnery prior to incubation whereas the other fed on natural food. This difference in the birds’ nutritional history allowed us to assess the effects of food on reproduction. Laying occurred between mid-April and early June, with a peak at the end of April or early May. Eggs were laid around noon. Adult females produced one clutch per year. Clutch size averaged 7.4 eggs (4–11). Incubation lasted 24–25 days. We observed a higher nesting success (67.7%) than reported for other eared pheasants. Pro- visioning had no significant effect on the timing of clutch initiation or nesting success, and a weak effect on egg size and clutch size (explaining 8.2% and 9.1% of the observed variation, respectively). These results were attributed to the observation that the unprovisioned birds had not experienced local food shortage before laying, despite spending more time feeding and less time resting than the provisioned birds. Nest-site selection by the pheasants was non-random with respect to environmental variables. Rock-cavities with an entrance aver- aging 0.32 m2 in size and not deeper than 1.5 m were greatly preferred as nest-sites. -

Rapanui Fowl
BREED FOCUS: RAPANUI FOWL By Christine Heinrichs and Kermit Blackwood (USA) All photos not individually credited are by either Michelle Tullis, Jeremy Yashar-Johnson, Martin Blendulf or Tamiym Lehoux Above: A View of the Monuments of Easter Island, Rapa Nui. Oil on panel, by William Hodges, 1870. On Easter Sunday, April 5, 1722, Dutch sea captain Jacob Roggeveen landed his ship the Tienhoven on an island the inhabitants, the Rapanui, called Te Pito o Te Henua, The Center of the World. Captain Roggeveen renamed it Easter Island. Right: Captain Jacob Roggeveen. By the time the Dutch arrived, Easter Island’s population was in decline. There were probably no more than about 2,000-3,000 people living on the island. Maximum population estimates of previous eras range from 4,000- 15,000. Theories differ as to the cause of their demise. Barbara A. West writes in the Encyclopedia of the Peoples of Asia and Oceania, “… the Rapanui experienced a tremendous upheaval in their social system brought about by a change in their island's ecology...” Jared Diamond (an American biogeographer) drew attention to the connection between ecological overexploitation and the downfall of society. In Easter Island we are describing a situation where wholesale ecological exploitation destroyed a civilization. They’d cut down every last trees making it impossible to so build seaworthy boats to sail away. As in other Polynesian cultures, the Rapanui people knew cannibalism. By the 18th century, the population of Easter Island were coping with staying warm from Antarctic winds in winter without sufficient clothing. Terry Hunt and Caro Lipo’s studies suggest that humans made permanent settlements on Easter Island around 1200 AD, and that the population never exceeded around 3,000 people. -

A Molecular Phylogeny of the Peacock-Pheasants (Galliformes: Polyplectron Spp.) Indicates Loss and Reduction of Ornamental Traits and Display Behaviours
Biological Journal of the Linnean Society (2001), 73: 187–198. With 3 figures doi:10.1006/bijl.2001.0536, available online at http://www.idealibrary.com on A molecular phylogeny of the peacock-pheasants (Galliformes: Polyplectron spp.) indicates loss and reduction of ornamental traits and display behaviours REBECCA T. KIMBALL1,2∗, EDWARD L. BRAUN1,3, J. DAVID LIGON1, VITTORIO LUCCHINI4 and ETTORE RANDI4 1Department of Biology, University of New Mexico, Albuquerque, NM 87131, USA 2Department of Evolution, Ecology and Organismal Biology, Ohio State University, Columbus, OH 43210, USA 3Department of Plant Biology, Ohio State University, Columbus, OH 43210, USA 4Istituto Nazionale per la Fauna Selvatica, via Ca` Fornacetta 9, 40064 Ozzano dell’Emilia (BO), Italy Received 4 September 2000; accepted for publication 3 March 2001 The South-east Asian pheasant genus Polyplectron is comprised of six or seven species which are characterized by ocelli (ornamental eye-spots) in all but one species, though the sizes and distribution of ocelli vary among species. All Polyplectron species have lateral displays, but species with ocelli also display frontally to females, with feathers held erect and spread to clearly display the ocelli. The two least ornamented Polyplectron species, one of which completely lacks ocelli, have been considered the primitive members of the genus, implying that ocelli are derived. We examined this hypothesis phylogenetically using complete mitochondrial cytochrome b and control region sequences, as well as sequences from intron G in the nuclear ovomucoid gene, and found that the two least ornamented species are in fact the most recently evolved. Thus, the absence and reduction of ocelli and other ornamental traits in Polyplectronare recent losses. -

Hybridization & Zoogeographic Patterns in Pheasants
University of Nebraska - Lincoln DigitalCommons@University of Nebraska - Lincoln Paul Johnsgard Collection Papers in the Biological Sciences 1983 Hybridization & Zoogeographic Patterns in Pheasants Paul A. Johnsgard University of Nebraska-Lincoln, [email protected] Follow this and additional works at: https://digitalcommons.unl.edu/johnsgard Part of the Ornithology Commons Johnsgard, Paul A., "Hybridization & Zoogeographic Patterns in Pheasants" (1983). Paul Johnsgard Collection. 17. https://digitalcommons.unl.edu/johnsgard/17 This Article is brought to you for free and open access by the Papers in the Biological Sciences at DigitalCommons@University of Nebraska - Lincoln. It has been accepted for inclusion in Paul Johnsgard Collection by an authorized administrator of DigitalCommons@University of Nebraska - Lincoln. HYBRIDIZATION & ZOOGEOGRAPHIC PATTERNS IN PHEASANTS PAUL A. JOHNSGARD The purpose of this paper is to infonn members of the W.P.A. of an unusual scientific use of the extent and significance of hybridization among pheasants (tribe Phasianini in the proposed classification of Johnsgard~ 1973). This has occasionally occurred naturally, as for example between such locally sympatric species pairs as the kalij (Lophura leucol11elana) and the silver pheasant (L. nycthelnera), but usually occurs "'accidentally" in captive birds, especially in the absence of conspecific mates. Rarely has it been specifically planned for scientific purposes, such as for obtaining genetic, morphological, or biochemical information on hybrid haemoglobins (Brush. 1967), trans ferins (Crozier, 1967), or immunoelectrophoretic comparisons of blood sera (Sato, Ishi and HiraI, 1967). The literature has been summarized by Gray (1958), Delacour (1977), and Rutgers and Norris (1970). Some of these alleged hybrids, especially those not involving other Galliformes, were inadequately doculnented, and in a few cases such as a supposed hybrid between domestic fowl (Gallus gal/us) and the lyrebird (Menura novaehollandiae) can be discounted. -

Assessment of Hematological Indices of Indian Peafowl (Pavo Cristatus) Kept at Wildlife Breeding Centre, Gatwala, Faisalabad, Pakistan
Journal of Zoological Research Volume 4, Issue 1, 2020, PP 29-33 ISSN 2637-5575 Assessment of Hematological Indices of Indian Peafowl (Pavo Cristatus) Kept at Wildlife Breeding Centre, Gatwala, Faisalabad, Pakistan Misbah Sarwar1* Zahid Ali1, and Muhammad Bilal Chaudhary2 1Punjab Wildlife Research Centre, Gatwala, Faisalabad Department of Wildlife & Parks, Punjab Pakistan 2Department of Zoology, GC University, Faisalabad, Pakistan *Corresponding Author: Misbah Sarwar, Punjab Wildlife Research Centre, Gatwala, Faisalabad Department of Wildlife & Parks, Punjab Pakistan. Email: [email protected] ABSTRACT Indian peafowl (Pavo cristatus) or blue peafowl has been maintained in captivity since long where due to selective breeding, several color mutations/varieties have appeared of which white peafowl, black-shouldered peafowl and pied peafowl are common. Since, hematological analysis is crucial for clinical diagnosis of wild and captive avifauna, so we collected blood samples from healthy male blue peafowl, white peafowl and black- shouldered peafowl kept at Wildlife Breeding Centre, Gatwala, Faisalabad and compared erythrocyte and leucocyte indices among them. Our results indicated that blood physiological values for MO (%), Hgb, HCT, MCH and MCHC were significantly different (P<0.05) between blue peafowl and white peafowl whereas MCV and RDW were significantly different (P<0.05) between blue peafowl and black-shouldered peafowl. The comparison of hematological parameters between white peafowl and black-shouldered peafowl showed that GR(%), RBC, HCT, MCV and MCHC differ significantly (P<0.05) between the two varieties. Our results support the studies indicating high quality color patterns reflect increased resistance and immunity to pathogens. Keywords: Indian peafowl, Color Mutations, hematology. INTRODUCTION 2005; Takahashi and Hasegawa, 2008; Harikrishnan et al., 2010, Naseer et al., 2017). -

Swierstra's Francolin Francolinus Swierstrai
abcbul 28-070718.qxp 7/18/2007 2:05 PM Page 175 Swierstra’s Francolin Francolinus swierstrai: a bibliography and summary of specimens Michael S. L. Mills Le Francolin de Swierstra Francolinus swierstrai: bibliographie et catalogue des spécimens. Le Francolin de Swierstra Francolinus swierstrai, endémique aux montagnes de l’Angola occidental, est considérée comme une espèce menacée (avec le statut de ‘Vulnérable’). En l’absence d’obser- vations entre 1971 et 2005, nous connaissons très peu de choses sur cette espèce. Cette note résume l’information disponible, basée sur 19 spécimens récoltés de 1907 à 1971, et présente une bibliographie complète, dans l’espoir d’encourager plus de recherches sur l’espèce. Summary. Swierstra’s Francolin Francolinus swierstrai is the only threatened bird endemic to the montane region of Western Angola. With no sightings between 1971 and August 2005, knowl- edge of this species is very poor. This note presents a summary of available information, based on 19 specimens collected between 1907 and 1971, and provides a complete bibliography, in order to encourage further work on this high-priority species. wierstra’s Francolin Francolinus swierstrai (or SSwierstra’s Spurfowl Pternistis swierstrai) was last recorded in February 1971 (Pinto 1983), until its rediscovery at Mt Moco in August 2005 (Mills & Dean in prep.). This Vulnerable species (BirdLife International 2000, 2004) is the only threatened bird endemic to montane western Angola, an area of critical importance for biodiver- sity conservation (Bibby et al. 1992, Stattersfield et al. 1998). Swierstra’s Francolin has a highly fragmented range of c.18,500 km2 and is suspected, despite the complete lack of sightings for more than 30 years, to have a declining population estimated at 2,500–9,999.