Signal, Noise, and Detection Limits in Mass Spectrometry
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Sensitive Determination of PAH-DNA Adducts
View metadata, citation and similar papers at core.ac.uk brought to you by CORE provided by Elsevier - Publisher Connector Matrix Design for Matrix-Assisted Laser Desorption Ionization: Sensitive Determination of PAH-DNA Adducts M. George, J. M. Y. Wellemans, R. L. Cerny, and M. L. Gross* Midwest Center for Mass Spectrometry, Department of Chemistry, University of Nebraska-Lincoln, Lincoln, Nebraska USA K. Li and E. L. Cavalieri Eppley institute for Research in Cancer and Allied Diseases, University of Nebraska Medical Center, 600 South 42nd Street, Omaha, Nebraska USA Two matrices, 4-phenyl-cw-cyanocinnamic acid (ICC) and 4-benzyloxy-cu-cyanocinnamic acid (BCC), were identified for the determination of polycyclic aromatic hydrocarbon @‘AH) adducts of DNA bases by matrix-assisted laser desorption ionization. These matrices were designed based on the concept that the matrix and the analyte should have structural similarities. PCC is a good matrix for the desorption of not only PA&modified DNA bases, but also PAHs themselves and their rnetabolites. Detections can be made at the femtomolar level. (1 Am Sot Mass Spectrum 1994, 5, 1021-1025) he success of matrix-assisted laser desorption quire sensitive detection methods at the low picomole ionization (MALDI) for the sensitive detection of and mid-femtomole level, respectively [6]. T an analyte depends on the nature of the matrix. One method for detection that also gives structural Rational design of a matrix requires understanding of information is fast-atom bombardment (FAB) coupled the mechanism of MALDI. The mechanism is not fully with tandem mass spectrometry [6-81. Unfortunately, understood. Nevertheless, empirical criteria for matrix the combination lacks sensitivity because there is in selection have been suggested 111. -

Bernoulli-Gaussian Distribution with Memory As a Model for Power Line Communication Noise Victor Fernandes, Weiler A
XXXV SIMPOSIO´ BRASILEIRO DE TELECOMUNICAC¸ OES˜ E PROCESSAMENTO DE SINAIS - SBrT2017, 3-6 DE SETEMBRO DE 2017, SAO˜ PEDRO, SP Bernoulli-Gaussian Distribution with Memory as a Model for Power Line Communication Noise Victor Fernandes, Weiler A. Finamore, Mois´es V. Ribeiro, Ninoslav Marina, and Jovan Karamachoski Abstract— The adoption of Additive Bernoulli-Gaussian Noise very useful to come up with a as simple as possible models (ABGN) as an additive noise model for Power Line Commu- of PLC noise, similar to the additive white Gaussian noise nication (PLC) systems is the subject of this paper. ABGN is a (AWGN) model. model that for a fraction of time is a low power noise and for the remaining time is a high power (impulsive) noise. In this context, In this regard, we introduce the definition of a simple and we investigate the usefulness of the ABGN as a model for the useful mathematical model for the noise perturbing the data additive noise in PLC systems, using samples of noise registered transmission over PLC channel that would be a helpful tool. during a measurement campaign. A general procedure to find the With this goal in mind, an investigation of the usefulness of model parameters, namely, the noise power and the impulsiveness what we call additive Bernoulli-Gaussian noise (ABGN) as a factor is presented. A strategy to assess the noise memory, using LDPC codes, is also examined and we came to the conclusion that model for the noise perturbing the digital communication over ABGN with memory is a consistent model that can be used as for PLC channel is discussed. -
EP Signal to Noise in Emp 2
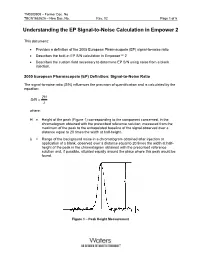
TN0000909 – Former Doc. No. TECN1852625 – New Doc. No. Rev. 02 Page 1 of 5 Understanding the EP Signal-to-Noise Calculation in Empower 2 This document: • Provides a definition of the 2005 European Pharmacopeia (EP) signal-to-noise ratio • Describes the built-in EP S/N calculation in Empower™ 2 • Describes the custom field necessary to determine EP S/N using noise from a blank injection. 2005 European Pharmacopeia (EP) Definition: Signal-to-Noise Ratio The signal-to-noise ratio (S/N) influences the precision of quantification and is calculated by the equation: 2H S/N = λ where: H = Height of the peak (Figure 1) corresponding to the component concerned, in the chromatogram obtained with the prescribed reference solution, measured from the maximum of the peak to the extrapolated baseline of the signal observed over a distance equal to 20 times the width at half-height. λ = Range of the background noise in a chromatogram obtained after injection or application of a blank, observed over a distance equal to 20 times the width at half- height of the peak in the chromatogram obtained with the prescribed reference solution and, if possible, situated equally around the place where this peak would be found. Figure 1 – Peak Height Measurement TN0000909 – Former Doc. No. TECN1852625 – New Doc. No. Rev. 02 Page 2 of 5 In Empower 2 software, the EP Signal-to-Noise (EP S/N) is determined in an automated fashion and does not require a blank injection. The intention of this calculation is to preserve the sense of the EP calculation without requiring a separate blank injection. -

Measurement and Estimation of the Mode Partition Coefficient K
Measurement and Estimation of the Mode Partition Coefficient k Rick Pimpinella and Jose Castro IEEE P802.3bm 40 Gb/s and 100 Gb/s Fiber Optic Task Force November 2012, San Antonio, TX Background & Objective Background: Mode Partition Noise in MMF channel links is caused by pulse-to-pulse power fluctuations among VCSEL modes and differential delay due to dispersion in the fiber (Power independent penalty) “k” is an index used to describe the degree of mode fluctuations and takes on a value between 0 and 1, called the mode partition coefficient [1,2] Currently the IEEE link model assumes k = 0.3 It has been discussed that a new link model requires validation of k The measurement & estimation of k is challenging due to several conditions: Low sensitivity of detectors at 850nm The presence of additional noise components in VCSEL-MMF channels (RIN, MN, Jitter – intensity fluctuations, reflection noise, thermal noise, …) Differences in VCSEL designs Objective: Provide an experimental estimate of the value of k for VCSELs Additional work in progress 2 MPN Theory Originally derived for Fabry-Perot lasers and SMF [3] Assumptions: Total power of all modes carried by each pulse is constant Power fluctuations among modes are anti-correlated Mechanism: When modes (different wavelengths) travel at the same speed their fluctuations remain anti-correlated and the resultant pulse-to-pulse noise is zero. When modes travel in a dispersive medium the modes undergo different delays resulting in pulse distortions and a noise penalty. Example: Two VCSEL modes transmitted in a SMF undergoing chromatic dispersion: START END Optical Waveform @ Detector START t END With mode power fluctuations Decision Region Shorter Wavelength Time (arrival) t slope DL Time (arrival) Longer Wavelength Time (arrival) 3 Example of Intensity Fluctuation among VCSEL modes MPN can be observed using an Optical Spectrum Analyzer (OSA). -

Introduction to Noise Measurements on Business Machines (Bo0125)
BO 0125-11 Introduction to Noise Measurements on Business Machines by Roger Upton, B.Sc. Introduction In recent years, manufacturers of DIN 45 635 pt.19 ization committees in the pursuit of business machines, that is computing ANSI Si.29, (soon to be superced- harmonization, meaning that the stan- and office machines, have come under ed by ANSI S12.10) dards agree with each other in the increasing pressure to provide noise ECMA 74 bulk of their requirements. This appli- data on their products. For instance, it ISO/DIS 7779 cation note first discusses some acous- is becoming increasingly common to tic measurement parameters, and then see noise levels advertised, particular- With four separate standards in ex- the measurements required by the ly for printers. A number of standards istence, one might expect some con- various standards. Finally, it describes have been written governing how such siderable divergence in their require- some Briiel & Kjaer measurement sys- noise measurements should be made, ments. However, there has been close terns capable of making the required namely: contact between the various standard- measurements. Introduction to Noise Measurement Parameters The standards referred to in the previous section call for measure• ments of sound power and sound pres• sure. These measurements can then be used in a variety of ways, such as noise labelling, prediction of installation noise levels, comparison of noise emis• sions of products, and production con• trol. Sound power and sound pressure are fundamentally different quanti• ties. Sound power is a measure of the ability of a device to make noise. It is independent of the acoustic environ• ment. -

Background Noise, Noise Sensitivity, and Attitudes Towards Neighbours, and a Subjective Experiment Using a Rubber Ball Impact Sound
International Journal of Environmental Research and Public Health Article Background Noise, Noise Sensitivity, and Attitudes towards Neighbours, and a Subjective Experiment Using a Rubber Ball Impact Sound Jeongho Jeong Fire Insurers Laboratories of Korea, 1030 Gyeongchungdae-ro, Yeoju-si 12661, Korea; [email protected]; Tel.: +82-31-887-6737 Abstract: When children run and jump or adults walk indoors, the impact sounds conveyed to neighbouring households have relatively high energy in low-frequency bands. The experience of and response to low-frequency floor impact sounds can differ depending on factors such as the duration of exposure, the listener’s noise sensitivity, and the level of background noise in housing complexes. In order to study responses to actual floor impact sounds, it is necessary to investigate how the response is affected by changes in the background noise and differences in the response when focusing on other tasks. In this study, the author presented subjects with a rubber ball impact sound recorded from different apartment buildings and housings and investigated the subjects’ responses to varying levels of background noise and when they were assigned tasks to change their level of attention on the presented sound. The subjects’ noise sensitivity and response to their neighbours were also compared. The results of the subjective experiment showed differences in the subjective responses depending on the level of background noise, and high intensity rubber ball impact sounds were associated with larger subjective responses. In addition, when subjects were performing a Citation: Jeong, J. Background Noise, task like browsing the internet, they attended less to the rubber ball impact sound, showing a less Noise Sensitivity, and Attitudes towards Neighbours, and a Subjective sensitive response to the same intensity of impact sound. -

The Effects of Background Noise on Distortion Product Otoacoustic Emissions and Hearing Screenings Amy E
Louisiana Tech University Louisiana Tech Digital Commons Doctoral Dissertations Graduate School Spring 2012 The effects of background noise on distortion product otoacoustic emissions and hearing screenings Amy E. Hollowell Louisiana Tech University Follow this and additional works at: https://digitalcommons.latech.edu/dissertations Part of the Speech Pathology and Audiology Commons Recommended Citation Hollowell, Amy E., "" (2012). Dissertation. 348. https://digitalcommons.latech.edu/dissertations/348 This Dissertation is brought to you for free and open access by the Graduate School at Louisiana Tech Digital Commons. It has been accepted for inclusion in Doctoral Dissertations by an authorized administrator of Louisiana Tech Digital Commons. For more information, please contact [email protected]. THE EFFECTS OF BACKGROUND NOISE ON DISTORTION PRODUCT OTOACOUSTIC EMISSIONS AND HEARING SCREENINGS by Amy E. Hollowell, B.S. A Dissertation Presented in Partial Fulfillment of the Requirements of the Degree Doctor of Audiology COLLEGE OF LIBERAL ARTS LOUISIANA TECH UNIVERSITY May 2012 UMI Number: 3515927 All rights reserved INFORMATION TO ALL USERS The quality of this reproduction is dependent upon the quality of the copy submitted. In the unlikely event that the author did not send a complete manuscript and there are missing pages, these will be noted. Also, if material had to be removed, a note will indicate the deletion. DiygrMution UMI 3515927 Published by ProQuest LLC 2012. Copyright in the Dissertation held by the Author. Microform Edition © ProQuest LLC. All rights reserved. This work is protected against unauthorized copying under Title 17, United States Code. ProQuest LLC 789 East Eisenhower Parkway P.O. Box 1346 Ann Arbor, Ml 48106-1346 LOUISIANA TECH UNIVERSITY THE GRADUATE SCHOOL 2/27/2012 Date We hereby recommend that the dissertation prepared under our supervision by Amy E. -

Effects of Aircraft Noise Exposure on Heart Rate During Sleep in the Population Living Near Airports
International Journal of Environmental Research and Public Health Article Effects of Aircraft Noise Exposure on Heart Rate during Sleep in the Population Living Near Airports Ali-Mohamed Nassur 1,*, Damien Léger 2, Marie Lefèvre 1, Maxime Elbaz 2, Fanny Mietlicki 3, Philippe Nguyen 3, Carlos Ribeiro 3, Matthieu Sineau 3, Bernard Laumon 4 and Anne-Sophie Evrard 1 1 Université Lyon, Université Claude Bernard Lyon1, IFSTTAR, UMRESTTE, UMR T_9405, F-69675 Bron, France; [email protected] (M.L.); [email protected] (A.-S.E.) 2 Université Paris Descartes, APHP, Hôtel-Dieu de Paris, Centre du Sommeil et de la Vigilance et EA 7330 VIFASOM, 75004 Paris, France; [email protected] (D.L.); [email protected] (M.E.) 3 Bruitparif, the Center for Technical Assessment of the Noise Environment in the Île-de-France Region of France, 93200 Saint-Denis, France; [email protected] (F.M.); [email protected] (P.N.); [email protected] (C.R.); [email protected] (M.S.) 4 IFSTTAR, Transport, Health and Safety Department, F-69675 Bron, France; [email protected] * Correspondence: [email protected] Received: 30 October 2018; Accepted: 15 January 2019; Published: 18 January 2019 Abstract: Background Noise in the vicinity of airports is a public health problem. Many laboratory studies have shown that heart rate is altered during sleep after exposure to road or railway noise. Fewer studies have looked at the effects of exposure to aircraft noise on heart rate during sleep in populations living near airports. Objective The aim of this study was to investigate the relationship between the sound pressure level (SPL) of aircraft noise and heart rate during sleep in populations living near airports in France. -

Audio Processing Laboratory
AC 2010-1594: A GRADUATE LEVEL COURSE: AUDIO PROCESSING LABORATORY Buket Barkana, University of Bridgeport Page 15.35.1 Page © American Society for Engineering Education, 2010 A Graduate Level Course: Audio Processing Laboratory Abstract Audio signal processing is a part of the digital signal processing (DSP) field in science and engineering that has developed rapidly over the past years. Expertise in audio signal processing - including speech signal processing- is becoming increasingly important for working engineers from diverse disciplines. Audio signals are stored, processed, and transmitted using digital techniques. Creating these technologies requires engineers that understand real-time application issues in the related areas. With this motivation, we designed a graduate level laboratory course which is Audio Processing Laboratory in the electrical engineering department in our school two years ago. This paper presents the status of the audio processing laboratory course in our school. We report the challenges that we have faced during the development of this course in our school and discuss the instructor’s and students’ assessments and recommendations in this real-time signal-processing laboratory course. 1. Introduction Many DSP laboratory courses are developed for undergraduate and graduate level engineering education. These courses mostly focus on the general signal processing techniques such as quantization, filter design, Fast Fourier Transformation (FFT), and spectral analysis [1-3]. Because of the increasing popularity of Web-based education and the advancements in streaming media applications, several web-based DSP Laboratory courses have been designed for distance education [4-8]. An internet-based signal processing laboratory that provides hands-on learning experiences in distributed learning environments has been developed by Spanias et.al[6] . -

Why Use Signal-To-Noise As a Measure of MS Performance When It Is Often Meaningless?
Why use Signal-To-Noise as a Measure of MS Performance When it is Often Meaningless? Technical Overview Authors Abstract Greg Wells, Harry Prest, and The signal-to-noise of a chromatographic peak determined from a single measurement Charles William Russ IV, has served as a convenient figure of merit used to compare the performance of two Agilent Technologies, Inc. different MS systems. The evolution in the design of mass spectrometry instrumenta- 2850 Centerville Road tion has resulted in very low noise systems that have made the comparison of perfor- Wilmington, DE 19809-1610 mance based upon signal-to-noise increasingly difficult, and in some modes of opera- USA tion impossible. This is especially true when using ultra-low noise modes such as high resolution mass spectrometry or tandem MS; where there are often no ions in the background and the noise is essentially zero. This occurs when analyzing clean stan- dards used to establish the instrument specifications. Statistical methodology com- monly used to establish method detection limits for trace analysis in complex matri- ces is a means of characterizing instrument performance that is rigorously valid for both high and low background noise conditions. Instrument manufacturers should start to provide customers an alternative performance metric in the form of instrument detection limits based on relative the standard deviation of replicate injections to allow analysts a practical means of evaluating an MS system. Introduction of polysiloxane at m/z 281 increased baseline noise for HCB at m/z 282). Signal-to-noise ratio (SNR) has been a primary standard for Improvement in instrument design and changes to test com- comparing the performance of chromatography systems includ- pounds were accompanied by a number of different ing GC/MS and LC/MS. -

Robust Signal-To-Noise Ratio Estimation Based on Waveform Amplitude Distribution Analysis
Robust Signal-to-Noise Ratio Estimation Based on Waveform Amplitude Distribution Analysis Chanwoo Kim, Richard M. Stern Department of Electrical and Computer Engineering and Language Technologies Institute Carnegie Mellon University, Pittsburgh, PA 15213 {chanwook, rms}@cs.cmu.edu speech and background noise are independent, (2) clean Abstract speech follows a gamma distribution with a fixed shaping In this paper, we introduce a new algorithm for estimating the parameter, and (3) the background noise has a Gaussian signal-to-noise ratio (SNR) of speech signals, called WADA- distribution. Based on these assumptions, it can be seen that if SNR (Waveform Amplitude Distribution Analysis). In this we model noise-corrupted speech at an unknown SNR using algorithm we assume that the amplitude distribution of clean the Gamma distribution, the value of the shaping parameter speech can be approximated by the Gamma distribution with obtained using maximum likelihood (ML) estimation depends a shaping parameter of 0.4, and that an additive noise signal is uniquely on the SNR. Gaussian. Based on this assumption, we can estimate the SNR While we assume that the background noise can be by examining the amplitude distribution of the noise- assumed to be Gaussian, we will demonstrate that this corrupted speech. We evaluate the performance of the algorithm still provides better results than the NIST STNR WADA-SNR algorithm on databases corrupted by white algorithm, even in the presence of other types of maskers such noise, background music, and interfering speech. The as background music or interfering speech, where the WADA-SNR algorithm shows significantly less bias and less corrupting signal is clearly not Gaussian. -

Isoprene Photochemistry Over the Amazon Rainforest
Isoprene photochemistry over the Amazon rainforest Yingjun Liua, Joel Britob, Matthew R. Dorrisc, Jean C. Rivera-Riosc,d, Roger Secoe, Kelvin H. Batesf, Paulo Artaxob, Sergio Duvoisin Jr.g, Frank N. Keutscha,c,d, Saewung Kime, Allen H. Goldsteinh, Alex B. Guenthere,i, Antonio O. Manzij, Rodrigo A. F. Souzak, Stephen R. Springstonl, Thomas B. Watsonl, Karena A. McKinneya,1, and Scot T. Martina,m,1 aJohn A. Paulson School of Engineering and Applied Sciences, Harvard University, Cambridge, MA 02138; bDepartment of Applied Physics, University of São Paulo, São Paulo 05508, Brazil; cDepartment of Chemistry, University of Wisconsin-Madison, Madison, WI 53706; dDepartment of Chemistry and Chemical Biology, Harvard University, Cambridge, MA 02138; eDepartment of Earth System Science, University of California, Irvine, CA 92697; fDivision of Chemistry and Chemical Engineering, California Institute of Technology, Pasadena, CA 91125; gDepartment of Chemistry, Universidade do Estado do Amazonas, Manaus, AM 69050, Brazil; hDepartment of Environmental Science, Policy, and Management, University of California, Berkeley, CA 94720; iPacific Northwest National Laboratory, Richland, WA 99354; jInstituto Nacional de Pesquisas da Amazonia, Manaus, AM 69067, Brazil; kDepartment of Meteorology, Universidade do Estado do Amazonas, Manaus, AM 69050, Brazil; lDepartment of Environmental and Climate Sciences, Brookhaven National Laboratory, Upton, NY 11973; and mDepartment of Earth and Planetary Sciences, Harvard University, Cambridge, MA 02138 Edited by Mark H. Thiemens, University of California, San Diego, La Jolla, CA, and approved April 13, 2016 (received for review December 23, 2015) Isoprene photooxidation is a major driver of atmospheric chemistry forests like Amazonia where there are few anthropogenic NO over forested regions.