Wettability of Silicon, Silicon Dioxide, and Organosilicate Glass
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

What Is the Value of Water Contact Angle on Silicon?
materials Article What Is the Value of Water Contact Angle on Silicon? Paweł Bryk 1, Emil Korczeniewski 2, Grzegorz S. Szyma ´nski 2, Piotr Kowalczyk 3, Konrad Terpiłowski 4 and Artur P. Terzyk 2,* 1 Department of Chemistry, Chair of Theoretical Chemistry, Maria Curie-Skłodowska University, 20-031 Lublin, Poland; [email protected] 2 Faculty of Chemistry, Physicochemistry of Carbon Materials Research Group, Nicolaus Copernicus University in Toru´n,Gagarin Street 7, 87-100 Toru´n,Poland; [email protected] (E.K.); [email protected] (G.S.S.) 3 College of Science, Health, Engineering and Education, Murdoch University, Murdoch WA 6150, Australia; [email protected] 4 Department of Chemistry, Chair of Physical Chemistry of Interfacial Phenomena, Maria Curie-Skłodowska University, 20-031 Lublin, Poland; [email protected] * Correspondence: [email protected]; Tel.: +48-56-61-14-371 Received: 4 March 2020; Accepted: 26 March 2020; Published: 27 March 2020 Abstract: Silicon is a widely applied material and the wetting of silicon surface is an important phenomenon. However, contradictions in the literature appear considering the value of the water contact angle (WCA). The purpose of this study is to present a holistic experimental and theoretical approach to the WCA determination. To do this, we checked the chemical composition of the silicon (1,0,0) surface by using the X-ray photoelectron spectroscopy (XPS) method, and next this surface was purified using different cleaning methods. As it was proved that airborne hydrocarbons change a solid wetting properties the WCA values were measured in hydrocarbons atmosphere. Next, molecular dynamics (MD) simulations were performed to determine the mechanism of wetting in this atmosphere and to propose the force field parameters for silica wetting simulation. -

MAGNESIUM SILICATE, Synthetic
MAGNESIUM SILICATE, synthetic Prepared at the 74th JECFA (2011) and published in FAO Monographs 11 (2011) superseding specifications prepared at the 61st JECFA (2003), published in the Combined Compendium of Food Additive Specifications, FAO JECFA Monographs 1 (2005). An ADI ‘not specified’ was established at the 25th JECFA (1981). SYNONYMS INS No. 553(i) DEFINITION Magnesium silicate (synthetic) is manufactured by the precipitation reaction between sodium silicate and a soluble magnesium salt. The aqueous suspension of the precipitate is filtered and the collected solid washed, dried, classified for particle size and packaged. The finest material is intended for use as an anticaking agent and the coarser particles are for use as a filtering aid. The moisture content of the material meant for use as an anticaking agent is kept to less than 15%. Although magnesium silicate is of variable composition, the molar ratio of MgO to SiO2 is approximately 2:5. Chemical name Magnesium silicate C.A.S. number 1343-88-0 Assay Not less than 15% of MgO and not less than 67% of SiO2, calculated on the ignited basis. DESCRIPTION Very fine, white, odourless powder, free from grittiness FUNCTIONAL USES Anticaking agent, filtering aid CHARACTERISTICS IDENTIFICATION Solubility (Vol. 4) Insoluble in water pH (Vol. 4) 7.0-11.0 (1 in 10 slurry) Magnesium (Vol. 4) Mix about 0.5 g of the sample with 10 ml of dilute hydrochloric acid TS, filter, and neutralize the filtrate to litmus paper with ammonia TS. The neutralized filtrate gives a positive test for magnesium. Silicate Prepare a bead by fusing a few crystals of sodium ammonium phosphate on a platinum loop in the flame of a Bunsen burner. -
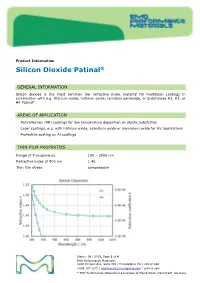
Sio2,Silicon Dioxide Patinal®Us
Product Information Silicon Dioxide Patinal ® GENERAL INFORMATION Silicon dioxide is the most common low refractive index material for multilayer coatings in combination with e.g. titanium oxide, hafnium oxide, tantalum pentoxide, or Substances H1, H2, or H4 Patinal . AREAS OF APPLICATION • Antireflection (AR) coatings for low temperature deposition on plastic substrates • Laser coatings, e.g. with hafnium oxide, scandium oxide or aluminium oxide for UV applications • Protective coating on Al-coatings THIN FILM PROPERTIES Range of Transparency 190 – 2000 nm Refractive index at 500 nm 1.46 Thin film stress compressive Status: 08 / 2019, Page 1 of 4 EMD Performance Materials* 1200 Intrepid Ave, Suite 300 / Philadelphia, PA / 19112 USA +888 367 3275 / [email protected] / patinal.com * EMD Performance Materials is a business of Merck KGaA, Darmstadt, Germany wavl / nm 200 250 350 500 650 800 1000 1250 n 1.551 1.531 1.487 1.470 1.463 1.461 1.458 1.457 k 7.1-04 2.6E-04 < 1E-4 < 1E-4 < 1E-4 < 1E-4 < 1E-4 < 1E-4 The resulting optical properties of the thin film are dependent on process conditions like deposition rate and substrate temperature. SiO 2 thin films exhibit an amorphous structure. High packing densities can be achieved by using IAD or a high substrate temperature. NOTES FOR EVAPORATION Evaporator source Electron beam evaporator Crucible / Boat Copper crucible Evaporation temperature 1800 – 2200 °C Deposition rate 0.4 – 1.2 nm/s Oxygen partial pressure none to ~5·10 -5 mbar IAD settings (Leybold APS) 130 – 150 V Bias Substrate temperature conventional RT to 350 °C IAD @ RT QCR-settings Density 2.648 g/cm³, z-ratio 1.0 The evaporation characteristics and the properties of the coating depend on the focusing of the electron beam. -

(Quartz) Silica, Sio2, Is a White Or Colorless Crystalline Compound
Silica (quartz) Silica, SiO2, is a white or colorless crystalline compound found mainly as quartz, sand, flint, and many other minerals. Silica is an important ingredient to manufacture a wide variety of materials. Quartz; Quartz is the most abundant silica mineral. Pure Quartz is colorless and transparent. It occurs in most igneous and practically all metamorphic and sedimentary rocks. It is used as a component of numerous industrial materials. Silicon (Si) has the atomic number 14 and is closely related to carbon. It is a relatively inert metalloid. Silicon is often used for microchips, glass, cement, and pottery. Silica is the most abundant mineral found in the crust of the earth. One of the most common uses of silica quarts is the manufacturer of glass. Silica is the fourteenth element on the periodic table. It can sometimes be found as the substance, quartz which is usually used in jewelry, test tubes, and when placed under pressure, generates an electrical charge. Quartz is the second most abundant mineral in the Earth's crust. It is a clear, glossy mineral with a hardness of 7 on the MOHS scale. Silica, Sa,is a component of glass and concrete. A form of Silica commonly known as quartz, Silica tetrahedra, is the second most common mineral in the earth's crust, it comes in many different forms. Silica is a compound of silicon and oxygen. Earth's outer crust contains 59% of this material. It has three major rock forms, which are quartz, tridymite, and cristobalite. Silica, commonly known in the form of quartz, is the dioxide form of silicon, SiO2. -

Silicon Oxide Etch Process
Silicon Oxide Etch Process To remove the areas of silicon dioxide unprotected by photoresist to remove SiO2 and expose the silicon underneath. Preparation and Precaution: 1. This operation should be started within 30 minutes of the completion of the hard bake of the photoresist. 2. Use Buffered Oxide Etch (BOE) solution (6 parts 40% NH4F and 1 part 49% HF). This reagent is found under the last acid fume hood in the Yellow Room. 2.1 Always use plastic beakers and graduated cylinders for etchants containing HF; HF WILL ETCH GLASS! 3. Know the oxide thickness on your substrate. Procedure: 1. Clean the substrate or wafer surface by rinsing with DI water; hold the sample with Teflon tweezers. 2. Before beginning the etching, calculate the time needed to etch through the oxide of known thickness using the known etch rate of the BOE solution; the etch rate at room temperature is ~700 A/min. 3. Use a stop watch to time the etching process. 4. Etch the wafer for about half of the estimated time. Remove your sample, rinse with DI H20. 5. If the silicon dioxide has not been completely etched away, you will observe a film of the DI water wetting or sheeting across the whole surface of the wafer. If the layer of SiO2 has been etched off, the DI water will not adhere to the back, except for a few isolated drops, and the wafer will appear dry and dull gray. This happens because SiO2 is hydrophylic as opposed to silicon which is hydrophobic. Observe the backside of the wafer to confirm this effect for a few seconds. -

Material Safety Data Sheet
Date of Issue: 21 September 2021 SAFETY DATA SHEET 1. SUBSTANCE AND SOURCE IDENTIFICATION Product Identifier SRM Number: 2557 SRM Name: Used Auto Catalyst (Monolith) Other Means of Identification: Not applicable. Recommended Use of This Material and Restrictions of Use This Standard Reference Material (SRM) is intended for use in evaluating chemical and instrumental methods for the analysis of platinum group metals and lead. A unit of SRM 2557 consists of a glass bottle containing 70 g of used auto catalyst as a fine powder [<74 µm (200 mesh)]. Company Information National Institute of Standards and Technology Standard Reference Materials Program 100 Bureau Drive, Stop 2300 Gaithersburg, Maryland 20899-2300 Telephone: 301-975-2200 Emergency Telephone ChemTrec: E-mail: [email protected] 1-800-424-9300 (North America) Website: http://www.nist.gov/srm +1-703-527-3887 (International) 2. HAZARDS IDENTIFICATION Classification Physical Hazard: Not classified. Health Hazard: Carcinogenicity Category 2 Label Elements Symbol Signal Word WARNING Hazard Statement(s): H351 Suspected of causing cancer (inhalation). Precautionary Statement(s): P201 Obtain special instructions before use. P202 Do not handle until all safety precautions have been read and understood. P281 Use personal protective equipment as required. P308+P313 If exposed or concerned: Get medical attention. P405 Store locked up. P501 Dispose of contents and container in accordance with local regulations. Hazards Not Otherwise Classified: Not applicable. Ingredients(s) with Unknown Acute Toxicity: Not applicable. SRM 2557 Page 1 of 7 3. COMPOSITION AND INFORMATION ON HAZARDOUS INGREDIENTS Substance: Used auto catalyst Other Designations: Recycled monolith auto catalyst This material is a complex mixture that has not been tested as a whole. -

Synthesis of Titanium Dioxide/Silicon Dioxide from Beach Sand As Photocatalyst for Cr and Pb Remediation
catalysts Article Synthesis of Titanium Dioxide/Silicon Dioxide from Beach Sand as Photocatalyst for Cr and Pb Remediation Diana Rakhmawaty Eddy 1,* , Soraya Nur Ishmah 1, Muhamad Diki Permana 1 and M. Lutfi Firdaus 2 1 Department of Chemistry, Faculty of Mathematics and Sciences, Universitas Padjadjaran, Jl. Raya Bandung-Sumedang km. 21 Jatinangor, Sumedang, Jawa Barat 45363, Indonesia; [email protected] (S.N.I.); [email protected] (M.D.P.) 2 Graduate School of Science Education, Bengkulu University, Jl. W.R. Supratman Kandang Limun, Bengkulu 38371, Indonesia; lutfi@unib.ac.id * Correspondence: [email protected]; Tel.: +62-8132-273-1173 Received: 30 September 2020; Accepted: 27 October 2020; Published: 29 October 2020 Abstract: Heavy metals are non-biodegradable and have a high toxicity effect on microorganisms which makes their presence in the environment extremely dangerous. The method of handling heavy metal waste by photocatalysis techniques using TiO2/SiO2 composite showed a good performance in reducing harmful pollutants. In this study, SiO2 from Bengkulu beach sand, Indonesia, was used as a support material for TiO2 photocatalyst to remove Cr(VI) and Pb(II). SiO2 was obtained through leaching techniques using NaOH as a solvent. The TiO2/SiO2 composite photocatalyst was synthesized using a solvothermal method at 130 ◦C and then characterized using X-ray diffraction (XRD), Fourier transform infrared spectroscopy (FTIR), scanning electron microscopy (SEM) and a particle size analyzer (PSA). Based on the XRD diffractogram, the synthesized TiO2 showed the anatase structure while the SiO2 showed the amorphous structure. The Ti–O–Si bond is defined in the infrared (IR) spectra, which indicates that the relationship between TiO2 and SiO2 is a chemical interaction. -

Chalcogenide Glass Materials for Integrated Infrared Photonics
Chalcogenide Glass Materials for Integrated Infrared Photonics by Vivek Singh B.S. Materials Science and Engineering Columbia University, 2009 Submitted to the Department of Materials Science and Engineering in Partial Fulfillment of the Requirements for the Degree of DOCTOR OF PHILOSOPHY at the MASSACHUSETTS INSTITUTE OF TECHNOLOGY September 2015 © 2015 Massachusetts Institute of Technology. All rights reserved. Signature of Author:_________________________________________ Department of Materials Science and Engineering July 20, 2015 Certified by:_____________________________________________ Lionel C. Kimerling Thomas Lord Professor of Materials Science and Engineering Thesis Supervisor Certified by:_____________________________________________ Anuradha M. Agarwal Principal Research Scientist, Materials Processing Center Thesis Supervisor Accepted by:_____________________________________________ Donald Sadoway Chair, Departmental Committee on Graduate Students 2 Chalcogenide Glass Materials for Integrated Infrared Photonics by Vivek Singh Submitted to the Department of Materials Science and Engineering on July 20, 2015 in Partial Fulfillment of the Requirements for the Degree of Doctor of Philosophy in Materials Science and Engineering Abstract Chalcogenide glasses (ChGs) are amorphous compounds containing the chalcogen elements (S, Se, Te) and exhibit wide infrared transparency windows. They are easy to synthesize in bulk and thin film forms and their compositional flexibility allows tuning of optical properties such as refractive index making them ideal for infrared photonics. We have studied the material attenuation in ChGs that arises due to the presence of impurities in the raw materials and established UV photolithography-based process flows that enable fabrication of chalcogenide glass waveguides and microresonators for near- and mid-IR wavelength ranges. Waveguides and optical resonators are key microphotonic elements for many on-chip applications such as telecommunications and chemical sensing. -

Effects of Absorption of Water and Other Reactive Species on the Fracture Properties of Organosilicate Glass Thin Films Ting Y
Effects of Absorption of Water and Other Reactive Species on the Fracture Properties of Organosilicate Glass Thin Films Ting Y. Tsui a*, Joost J. Vlassak b, Kelly Taylor a, Andrew J. McKerrow a, and Robert Kraft a a Silicon Technology Development, Texas Instruments Incorporated, Dallas, TX b Division of Engineering and Applied Sciences, Harvard University, Cambridge, MA 02138-2901 * Contact author: [email protected] Abstract This paper investigates the effects of the absorption of water and several other reactive species on the adhesion energy between organosilicate glass (OSG) thin films and various capping layers. The adhesion energy of these interfaces was measured by four-point bending tests conducted at room temperature in an ambient environment. The amount of absorbed reactant in the organosilicate glass films was controlled by exposing the capped organosilicate films to aqueous solutions for various periods of time. Results show that the initial degradation of the critical adhesion energy of the interfaces between OSG and tantalum, tantalum nitride (TaNx), silicon nitride (SiNx), and silicon dioxide (SiO2) capping layers is very fast, but that the adhesion energy reaches a saturation value thereafter. The reduced adhesion energy can be fully recovered to the “dry” value by baking the samples. A quantitative model based on Henry’s law is used to predict the adhesion degradation rate as a function of moisture exposure time. This model leads to a new and simple method for measuring the diffusion coefficient of water in OSG coatings or their interfaces. Introduction Silica-based dielectrics are often doped with carbon atoms and other organic molecules in order to reduce the permittivity of back-end-of-line (BEOL) inter-layer dielectrics. -

Chlorine for In-Situ Low-Temperature Silicon Surface Cleaning for Epitaxy
Chlorine Etching For In-Situ Low-Temperature Silicon Surface Cleaning For Epitaxy Applications K. H. Chung, and J. C. Sturm Princeton Institute of Science and Technology of Materials (PRISM) and Dept. of Electrical Engineering, Princeton University, Princeton, NJ, 08544, USA Chlorine in a nitrogen ambient is used to clean silicon surfaces of impurities by etching a thin layer from the surface prior to silicon epitaxial growth. Silicon etch rates of 1-10 nm/min could be achieved for temperatures from 525oC to 575oC. The etching of a thin layer of silicon from the surface is also capable of removing phosphorus from the surface, which conventionally is difficult to remove. Smooth surfaces and high epitaxial quality after chlorine etching are also demonstrated. Introduction Oxygen-free silicon surfaces are desired for high quality silicon epitaxy. Conventionally, to achieve oxygen-free and carbon-free surfaces, it is necessary to perform in-situ cleaning steps with temperatures in the range of 760oC to 850oC or higher to reduce oxygen levels to below the secondary ion mass spectrometry (SIMS) resolution limit (1)(2). Cleaning temperatures less than 700oC are desired, however, to minimize the diffusion of dopants in existing structures, such as would occur when using epitaxy for raised source/drain applications. In this work, we demonstrate the use of chlorine etching at 575oC to produce oxygen-free surfaces and substantially reduce interface phosphorus and carbon levels before epitaxy. Chlorine Thermal Etching Thermal etching of silicon in-situ in epitaxial reactors is typically done using hydrogen chloride (HCl) in a hydrogen ambient. However, the etch rates using this process are very low below 800oC. -

Extraction and Quantification of Silicon from Silica Sand Obtained from Zauma River, Zamfara State, Nigeria
View metadata, citation and similar papers at core.ac.uk brought to you by CORE provided by European Scientific Journal (European Scientific Institute) European Scientific Journal May 2013 edition vol.9, No.15 ISSN: 1857 – 7881 (Print) e - ISSN 1857- 7431 EXTRACTION AND QUANTIFICATION OF SILICON FROM SILICA SAND OBTAINED FROM ZAUMA RIVER, ZAMFARA STATE, NIGERIA Idrisa Wazamtu Email Department Of Pre-Nd Studies Ramat Polytechnic Maiduguri, Borno State, Nigeria Nasiru Abdullahi Sani Department Of Pure And Applied Chemistry, Usmanu Danfodiyo University Sokoto, Sokoto State, Nigeria Abba Kura Abdulsalam Chemical Technological Department, Standards Organization Of Nigeria, Lekki, Lagos State, Nigeria Umaru Abubakar Abdullahi G.S.S Kawu, Abuja Education Board Fct, Abuja, Nigeria Abstract This is a research on the extraction and quantification of Silicon from Silica Sand obtained from Zauma River in Zamfara state. The Silica Sand sample was removed from the water in Zauma River which was allowed to dry and heated in a heating mantle. Silicon and Silica was extracted from the sample. Quantitative analysis of the sample was then done under the following parameters; determination of loss on ignition, determination of CaO, determination of MgO, determination of Fe2O3, and determination of Al2O3.The results reveals that, the sample contains numerous cations in form of oxides and SiO2 and Si having the highest percentages of 81.5% and 77% respectively. Based on the findings some recommendations were given. Keywords: Silica and Silicon, Quantitative analysis, determination, Oxides, Elements Introduction Silica Sand is a brownish glassy with high reflection like broken glasses. It usually contains Silicon (Si) as the major element as its constituents and other cations (Ratson 2007). -

Toxicological Profile for Silica Released for Public Comment in 2017
Toxicological Profile for Silica September 2019 SILICA ii DISCLAIMER Use of trade names is for identification only and does not imply endorsement by the Agency for Toxic Substances and Disease Registry, the Public Health Service, or the U.S. Department of Health and Human Services. SILICA iii FOREWORD This toxicological profile is prepared in accordance with guidelines* developed by the Agency for Toxic Substances and Disease Registry (ATSDR) and the Environmental Protection Agency (EPA). The original guidelines were published in the Federal Register on April 17, 1987. Each profile will be revised and republished as necessary. The ATSDR toxicological profile succinctly characterizes the toxicologic and adverse health effects information for these toxic substances described therein. Each peer-reviewed profile identifies and reviews the key literature that describes a substance's toxicologic properties. Other pertinent literature is also presented, but is described in less detail than the key studies. The profile is not intended to be an exhaustive document; however, more comprehensive sources of specialty information are referenced. The focus of the profiles is on health and toxicologic information; therefore, each toxicological profile begins with a relevance to public health discussion which would allow a public health professional to make a real-time determination of whether the presence of a particular substance in the environment poses a potential threat to human health. The adequacy of information to determine a substance's health