Supplementary Appendix
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

FIP1L1 Gene FIP1 Like 1 (S
FIP1L1 gene FIP1 like 1 (S. cerevisiae) Normal Function The FIP1L1 gene provides instructions for making part of a protein complex named cleavage and polyadenylation specificity factor (CPSF). This complex of proteins plays an important role in processing molecules called messenger RNAs (mRNAs), which serve as the genetic blueprints for making proteins. The CPSF protein complex helps add a string of the RNA building block adenine to the mRNA, creating a polyadenine tail or poly(A) tail. The poly(A) tail is important for stability of the mRNA and for protein production from the blueprint. Health Conditions Related to Genetic Changes PDGFRA-associated chronic eosinophilic leukemia A deletion of genetic material from chromosome 4 brings together part of the FIP1L1 gene and part of another gene called PDGFRA, creating the FIP1L1-PDGFRA fusion gene. This mutation is a somatic mutation, which means it is acquired during a person's lifetime and is present only in certain cells. This fusion gene causes PDGFRA- associated chronic eosinophilic leukemia, which is a type of blood cell cancer characterized by an increased number of eosinophils, a type of white blood cell involved in allergic reactions. The FIP1L1-PDGFRA protein produced from the fusion gene has the function of the normal PDGFRA protein, which stimulates signaling pathways inside the cell that control many important cellular processes, such as cell growth and division (proliferation) and cell survival. Unlike the normal PDGFRA protein, however, the FIP1L1-PDGFRA protein is constantly turned on (constitutively activated), which means the cells are always receiving signals to proliferate. When the FIP1L1-PDGFRA fusion gene occurs in blood cell precursors, the growth of eosinophils (and occasionally other blood cells) is poorly controlled, leading to PDGFRA-associated chronic eosinophilic leukemia. -

R-Loop-Mediated Genome Instability in Mrna Cleavage and Polyadenylation Mutants
Downloaded from genesdev.cshlp.org on September 29, 2021 - Published by Cold Spring Harbor Laboratory Press R-loop-mediated genome instability in mRNA cleavage and polyadenylation mutants Peter C. Stirling,1,3 Yujia A. Chan,1,3 Sean W. Minaker,1,3 Maria J. Aristizabal,2 Irene Barrett,1 Payal Sipahimalani,1 Michael S. Kobor,2 and Philip Hieter1,4 1Michael Smith Laboratories, University of British Columbia, Vancouver, British Columbia V6T1Z4, Canada; 2Centre for Molecular Medicine and Therapeutics, Vancouver, British Columbia V5Z4H4, Canada Genome instability via RNA:DNA hybrid-mediated R loops has been observed in mutants involved in various aspects of transcription and RNA processing. The prevalence of this mechanism among essential chromosome instability (CIN) genes remains unclear. In a secondary screen for increased Rad52 foci in CIN mutants, representing ~25% of essential genes, we identified seven essential subunits of the mRNA cleavage and polyadenylation (mCP) machinery. Genome-wide analysis of fragile sites by chromatin immunoprecipitation (ChIP) and microarray (ChIP–chip) of phosphorylated H2A in these mutants supported a transcription-dependent mechanism of DNA damage characteristic of R loops. In parallel, we directly detected increased RNA:DNA hybrid formation in mCP mutants and demonstrated that CIN is suppressed by expression of the R-loop-degrading enzyme RNaseH. To investigate the conservation of CIN in mCP mutants, we focused on FIP1L1, the human ortholog of yeast FIP1, a conserved mCP component that is part of an oncogenic fusion in eosinophilic leukemia. We found that truncation fusions of yeast FIP1 analogous to those in cancer cause loss of function and that siRNA knockdown of FIP1L1 in human cells increases DNA damage and chromosome breakage. -

Hypereosinophilia in Granular Acute Bcell Lymphoblastic Leukemia With
bs_bs_banner Microscopic polyangiitis 543 and had been diagnosed with normocytic anemia.At that time, she References received treatment with an iron preparation because of a low 1 Ozen S, Ruperto N, Dillon MJ et al. EULAR/PReS endorsed con- serum iron level despite her normal ferritin level. We estimate that sensus criteria for the classification of childhood vasculitides. Ann. the patient already had mild alveolar hemorrhage at that time and Rheum. Dis. 2006; 65: 936–41. the bleeding later healed spontaneously. Among the cases of this 2 Vanoni F, Bettinelli A, Keller F, Bianchetti MG, Simonetti GD. disease reported from Japan, the disease tended to develop more Vasculitides associated with IgG antineutrophil cytoplasmic autoan- tibodies in childhood. Pediatr. Nephrol. 2010; 25: 205–12. frequently in the spring. In our patient, the disease developed twice 3 Kobayashi S, Inokuma S. Intrapulmonary hemorrhage in collagen- (both times in June), suggesting the possibility that infection vascular disease includes a spectrum of underlying conditions. triggered the disease onset. Although alveolar hemorrhage sub- Intern. Med. 2009; 48: 891–7. sided spontaneously in this case, lung fibrosis may develop in the 4 Hattori M, Kurayama H, Koitabashi Y. Antineutrophil cytoplasmic future if alveolar bleeding develops repeatedly and its diagnosis is autoantibody-associated glomerulonephritis in children. J. Am. Soc. Nephrol. 2001; 12: 1493–500. delayed. In the present case, treatment was started immediately 5 Peco-Antic A, Bonaci-Nikolic B, Basta-Jovanovic G et al. Child- after diagnosis and rapid aggravation of her renal function was hood microscopic polyangiitis associated with MPO-ANCA. prevented. When dealing with cases in whom unexplained nor- Pediatr. -

To Study Mutant P53 Gain of Function, Various Tumor-Derived P53 Mutants
Differential effects of mutant TAp63γ on transactivation of p53 and/or p63 responsive genes and their effects on global gene expression. A thesis submitted in partial fulfillment of the requirements for the degree of Master of Science By Shama K Khokhar M.Sc., Bilaspur University, 2004 B.Sc., Bhopal University, 2002 2007 1 COPYRIGHT SHAMA K KHOKHAR 2007 2 WRIGHT STATE UNIVERSITY SCHOOL OF GRADUATE STUDIES Date of Defense: 12-03-07 I HEREBY RECOMMEND THAT THE THESIS PREPARED UNDER MY SUPERVISION BY SHAMA KHAN KHOKHAR ENTITLED Differential effects of mutant TAp63γ on transactivation of p53 and/or p63 responsive genes and their effects on global gene expression BE ACCEPTED IN PARTIAL FULFILLMENT OF THE REQUIREMENTS FOR THE DEGREE OF Master of Science Madhavi P. Kadakia, Ph.D. Thesis Director Daniel Organisciak , Ph.D. Department Chair Committee on Final Examination Madhavi P. Kadakia, Ph.D. Steven J. Berberich, Ph.D. Michael Leffak, Ph.D. Joseph F. Thomas, Jr., Ph.D. Dean, School of Graduate Studies 3 Abstract Khokhar, Shama K. M.S., Department of Biochemistry and Molecular Biology, Wright State University, 2007 Differential effect of TAp63γ mutants on transactivation of p53 and/or p63 responsive genes and their effects on global gene expression. p63, a member of the p53 gene family, known to play a role in development, has more recently also been implicated in cancer progression. Mice lacking p63 exhibit severe developmental defects such as limb truncations, abnormal skin, and absence of hair follicles, teeth, and mammary glands. Germline missense mutations of p63 have been shown to be responsible for several human developmental syndromes including SHFM, EEC and ADULT syndromes and are associated with anomalies in the development of organs of epithelial origin. -

PRODUCT SPECIFICATION Prest Antigen FIP1L1 Product Datasheet
PrEST Antigen FIP1L1 Product Datasheet PrEST Antigen PRODUCT SPECIFICATION Product Name PrEST Antigen FIP1L1 Product Number APrEST79770 Gene Description factor interacting with PAPOLA and CPSF1 Alternative Gene DKFZp586K0717 Names Corresponding Anti-FIP1L1 (HPA037475) Antibodies Description Recombinant protein fragment of Human FIP1L1 Amino Acid Sequence Recombinant Protein Epitope Signature Tag (PrEST) antigen sequence: KANSSVGKWQDRYGRAESPDLRRLPGAIDVIGQTITISRVEGRRRANENS NIQVLSERSATEVDNNFSKPPPFFPPGAPPT Fusion Tag N-terminal His6ABP (ABP = Albumin Binding Protein derived from Streptococcal Protein G) Expression Host E. coli Purification IMAC purification Predicted MW 27 kDa including tags Usage Suitable as control in WB and preadsorption assays using indicated corresponding antibodies. Purity >80% by SDS-PAGE and Coomassie blue staining Buffer PBS and 1M Urea, pH 7.4. Unit Size 100 µl Concentration Lot dependent Storage Upon delivery store at -20°C. Avoid repeated freeze/thaw cycles. Notes Gently mix before use. Optimal concentrations and conditions for each application should be determined by the user. Product of Sweden. For research use only. Not intended for pharmaceutical development, diagnostic, therapeutic or any in vivo use. No products from Atlas Antibodies may be resold, modified for resale or used to manufacture commercial products without prior written approval from Atlas Antibodies AB. Warranty: The products supplied by Atlas Antibodies are warranted to meet stated product specifications and to conform to label descriptions when used and stored properly. Unless otherwise stated, this warranty is limited to one year from date of sales for products used, handled and stored according to Atlas Antibodies AB's instructions. Atlas Antibodies AB's sole liability is limited to replacement of the product or refund of the purchase price. -

Time Resolved Quantitative Phosphoproteomics Reveals Distinct Patterns of SHP2
bioRxiv preprint doi: https://doi.org/10.1101/598664; this version posted April 12, 2019. The copyright holder for this preprint (which was not certified by peer review) is the author/funder, who has granted bioRxiv a license to display the preprint in perpetuity. It is made available under aCC-BY-NC-ND 4.0 International license. Time resolved quantitative phosphoproteomics reveals distinct patterns of SHP2 dependence in EGFR signaling Vidyasiri Vemulapalli1,2, Lily Chylek3, Alison Erickson4, Jonathan LaRochelle1,2, Kartik Subramanian3, Morvarid Mohseni5, Matthew LaMarche5, Michael G. Acker5, Peter K. Sorger3, Steven P. Gygi4, and Stephen C. Blacklow1,2* 1Department of Cancer Biology, Dana-Farber Cancer Institute Boston, MA 02115, USA 2Department of Biological Chemistry & Molecular Pharmacology, Blavatnik Institute, Harvard Medical School, Boston, MA 02115, USA 3Laboratory of Systems Pharmacology, Harvard Medical School, Boston, MA 02115, USA 4Department of Cell Biology, Harvard Medical School, Boston, MA 02115, USA 5Novartis Institutes for Biomedical Research, Cambridge, MA, 02139, USA *To whom correspondence should be addressed: [email protected] bioRxiv preprint doi: https://doi.org/10.1101/598664; this version posted April 12, 2019. The copyright holder for this preprint (which was not certified by peer review) is the author/funder, who has granted bioRxiv a license to display the preprint in perpetuity. It is made available under aCC-BY-NC-ND 4.0 International license. Abstract SHP2 is a protein tyrosine phosphatase that normally potentiates intracellular signaling by growth factors, antigen receptors, and some cytokines; it is frequently mutated in childhood leukemias and other cancers. Here, we examine the role of SHP2 in the responses of breast cancer cells to EGF by monitoring phosphoproteome dynamics when SHP2 is allosterically inhibited by the small molecule SHP099. -
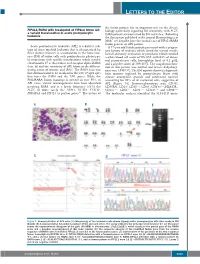
Letters to the Editor
LETTERS TO THE EDITOR the fusion partner has an important role on the disease FIP1L1/RARA with breakpoint at FIP1L1 intron 13: biology particularly regarding RA sensitivity, with PLZF- a variant translocation in acute promyelocytic 4 leukemia RARA patients characterized by RA resistance. Following the description published in the journal Haematologica in 2008, 5 we describe here the second case of FIP1L1/RARA fusion gene in an APL patient. Acute promyelocytic leukemia (APL) is a distinct sub - A 77-year old female patient presented with a progres - type of acute myeloid leukemia that is characterized by sive history of asthenia which lasted for several weeks. three distinct features: i) accumulation in the bone mar - Initial laboratory evaluation of peripheral blood revealed row (BM) of tumor cells with promyelocytic phenotype; a white blood cell count of 59 ¥10 9/L with 84% of abnor - ii) association with specific translocations which involve mal promyelocyte cells, hemoglobin level of 9.2 g/dL, chromosome 17 at the retinoic acid receptor alpha ( RARA ) and a platelet count of 109 ¥10 9/L. The coagulation func - locus ; iii) and the sensitivity of APL blasts to the differen - tion in this patient was normal and lactate dehydroge - 1 tiating action of retinoic acid (RA). The RARA locus was nase was 1,938 U/L. The BM aspirate showed a hypercel - first demonstrated to be involved in the t(15;17)(q22;q21) lular marrow replaced by promyelocyte blasts with that fuses the RARA and the PML genes. While the intense azurophilic granule and prominent nucleoli PML/RARA fusion transcript is present in over 95% of accounting for 93% of all nucleated cells, suggestive of APL cases, variant rearrangements have been identified APL (Figure 1A). -

The Regulatory Roles of Phosphatases in Cancer
Oncogene (2014) 33, 939–953 & 2014 Macmillan Publishers Limited All rights reserved 0950-9232/14 www.nature.com/onc REVIEW The regulatory roles of phosphatases in cancer J Stebbing1, LC Lit1, H Zhang, RS Darrington, O Melaiu, B Rudraraju and G Giamas The relevance of potentially reversible post-translational modifications required for controlling cellular processes in cancer is one of the most thriving arenas of cellular and molecular biology. Any alteration in the balanced equilibrium between kinases and phosphatases may result in development and progression of various diseases, including different types of cancer, though phosphatases are relatively under-studied. Loss of phosphatases such as PTEN (phosphatase and tensin homologue deleted on chromosome 10), a known tumour suppressor, across tumour types lends credence to the development of phosphatidylinositol 3--kinase inhibitors alongside the use of phosphatase expression as a biomarker, though phase 3 trial data are lacking. In this review, we give an updated report on phosphatase dysregulation linked to organ-specific malignancies. Oncogene (2014) 33, 939–953; doi:10.1038/onc.2013.80; published online 18 March 2013 Keywords: cancer; phosphatases; solid tumours GASTROINTESTINAL MALIGNANCIES abs in sera were significantly associated with poor survival in Oesophageal cancer advanced ESCC, suggesting that they may have a clinical utility in Loss of PTEN (phosphatase and tensin homologue deleted on ESCC screening and diagnosis.5 chromosome 10) expression in oesophageal cancer is frequent, Cao et al.6 investigated the role of protein tyrosine phosphatase, among other gene alterations characterizing this disease. Zhou non-receptor type 12 (PTPN12) in ESCC and showed that PTPN12 et al.1 found that overexpression of PTEN suppresses growth and protein expression is higher in normal para-cancerous tissues than induces apoptosis in oesophageal cancer cell lines, through in 20 ESCC tissues. -

PTPN11 Is the First Identified Proto-Oncogene That Encodes a Tyrosine Phosphatase
From www.bloodjournal.org by guest on July 4, 2016. For personal use only. Review article PTPN11 is the first identified proto-oncogene that encodes a tyrosine phosphatase Rebecca J. Chan1 and Gen-Sheng Feng2,3 1Department of Pediatrics, the Herman B. Wells Center for Pediatric Research, Indiana University School of Medicine, Indianapolis; 2Programs in Signal Transduction and Stem Cells & Regeneration, Burnham Institute for Medical Research, La Jolla, CA; 3Institute for Biomedical Research, Xiamen University, Xiamen, China Elucidation of the molecular mechanisms 2 Src-homology 2 (SH2) domains (Shp2). vation of the Ras-Erk pathway. This underlying carcinogenesis has benefited This tyrosine phosphatase was previ- progress represents another milestone in tremendously from the identification and ously shown to play an essential role in the leukemia/cancer research field and characterization of oncogenes and tumor normal hematopoiesis. More recently, so- provides a fresh view on the molecular suppressor genes. One new advance in matic missense PTPN11 gain-of-function mechanisms underlying cell transforma- this field is the identification of PTPN11 mutations have been detected in leuke- tion. (Blood. 2007;109:862-867) as the first proto-oncogene that encodes mias and rarely in solid tumors, and have a cytoplasmic tyrosine phosphatase with been found to induce aberrant hyperacti- © 2007 by The American Society of Hematology Introduction Leukemia and other types of cancer continue to be a leading cause tumor suppressor activity when overexpressed in vitro, and Ptprj of death in the United States, and biomedical scientists sorely note maps to the mouse colon cancer susceptibility locus,3 implicating that victories against cancer remain unacceptably rare. -

Mutational Landscape and Clinical Outcome of Patients with De Novo Acute Myeloid Leukemia and Rearrangements Involving 11Q23/KMT2A
Mutational landscape and clinical outcome of patients with de novo acute myeloid leukemia and rearrangements involving 11q23/KMT2A Marius Billa,1,2, Krzysztof Mrózeka,1,2, Jessica Kohlschmidta,b, Ann-Kathrin Eisfelda,c, Christopher J. Walkera, Deedra Nicoleta,b, Dimitrios Papaioannoua, James S. Blachlya,c, Shelley Orwicka,c, Andrew J. Carrolld, Jonathan E. Kolitze, Bayard L. Powellf, Richard M. Stoneg, Albert de la Chapelleh,i,2, John C. Byrda,c, and Clara D. Bloomfielda,c aThe Ohio State University Comprehensive Cancer Center, Columbus, OH 43210; bAlliance for Clinical Trials in Oncology Statistics and Data Center, The Ohio State University Comprehensive Cancer Center, Columbus, OH 43210; cDivision of Hematology, Department of Internal Medicine, The Ohio State University Comprehensive Cancer Center, Columbus, OH 43210; dDepartment of Genetics, University of Alabama at Birmingham, Birmingham, AL 35294; eNorthwell Health Cancer Institute, Zucker School of Medicine at Hofstra/Northwell, Lake Success, NY 11042; fDepartment of Internal Medicine, Section on Hematology & Oncology, Wake Forest Baptist Comprehensive Cancer Center, Winston-Salem, NC 27157; gDepartment of Medical Oncology, Dana-Farber/Partners Cancer Care, Boston, MA 02215; hHuman Cancer Genetics Program, Comprehensive Cancer Center, The Ohio State University, Columbus, OH 43210; and iDepartment of Cancer Biology and Genetics, Comprehensive Cancer Center, The Ohio State University, Columbus, OH 43210 Contributed by Albert de la Chapelle, August 28, 2020 (sent for review July 17, 2020; reviewed by Anne Hagemeijer and Stefan Klaus Bohlander) Balanced rearrangements involving the KMT2A gene, located at patterns that include high expression of HOXA genes and thereby 11q23, are among the most frequent chromosome aberrations in contribute to leukemogenesis (14–16). -

ORIGINAL ARTICLE the Severity of FIP1L1–PDGFRA-Positive Chronic
Leukemia (2007) 21, 2428–2432 & 2007 Nature Publishing Group All rights reserved 0887-6924/07 $30.00 www.nature.com/leu ORIGINAL ARTICLE The severity of FIP1L1–PDGFRA-positive chronic eosinophilic leukaemia is associated with polymorphic variation at the IL5RA locus S Burgstaller1, S Kreil1, K Waghorn1, G Metzgeroth2, C Preudhomme3, K Zoi4, H White1, D Cilloni5, C Zoi4, F Brito-Babapulle6, C Walz2, A Reiter2 and NCP Cross1 1Wessex Regional Genetics Laboratory, University of Southampton, Salisbury, UK; 2III Medizinische Universita¨tsklinik, Medizinische Fakulta¨t der Universita¨t Mannheim, Heidelberg, Germany; 3Laboratoire d’Hematologie A, CHU Lille, Lille, France; 4Foundation of Biomedical Research, Academy of Athens, Athens, Greece; 5Department of Clinical and Biological Sciences, University of Turin, Turin, Italy and 6Department of Haematology, Ealing Hospital, London, UK We have investigated the hypothesis that constitutional genetic Understanding of the myeloproliferative subtype of HES was variation in IL-5 signalling may be associated with the greatly advanced by the finding of the FIP1L1–PDGFRA fusion, development or severity of FIP1L1–PDGFRA-positive chronic formed by a cytogenetically cryptic 800 kb interstitial deletion at eosinophilic leukaemia (CEL) in humans. We genotyped six 2 single-nucleotide polymorphisms (SNP) within or close to the chromosome band 4q12. Although initially described in more IL5RA or IL5 genes in 82 patients with FIP1L1–PDGFRA- than 50% of cases with idiopathic HES, subsequent studies positive CEL plus, as controls, healthy individuals (n ¼ 100), estimated the prevalence to be lower at 3À17%.3–5 Detection of patients with FIP1L1–PDGFRA-negative eosinophilia (n ¼ 100) FIP1L1–PDGFRA by reverse transcription-PCR or fluorescent in or patients with chronic myeloid leukaemia (CML) (n ¼ 100). -

Genetic Alterations of Protein Tyrosine Phosphatases in Human Cancers
Oncogene (2015) 34, 3885–3894 © 2015 Macmillan Publishers Limited All rights reserved 0950-9232/15 www.nature.com/onc REVIEW Genetic alterations of protein tyrosine phosphatases in human cancers S Zhao1,2,3, D Sedwick3,4 and Z Wang2,3 Protein tyrosine phosphatases (PTPs) are enzymes that remove phosphate from tyrosine residues in proteins. Recent whole-exome sequencing of human cancer genomes reveals that many PTPs are frequently mutated in a variety of cancers. Among these mutated PTPs, PTP receptor T (PTPRT) appears to be the most frequently mutated PTP in human cancers. Beside PTPN11, which functions as an oncogene in leukemia, genetic and functional studies indicate that most of mutant PTPs are tumor suppressor genes. Identification of the substrates and corresponding kinases of the mutant PTPs may provide novel therapeutic targets for cancers harboring these mutant PTPs. Oncogene (2015) 34, 3885–3894; doi:10.1038/onc.2014.326; published online 29 September 2014 INTRODUCTION tyrosine/threonine-specific phosphatases. (4) Class IV PTPs include Protein tyrosine phosphorylation has a critical role in virtually all four Drosophila Eya homologs (Eya1, Eya2, Eya3 and Eya4), which human cellular processes that are involved in oncogenesis.1 can dephosphorylate both tyrosine and serine residues. Protein tyrosine phosphorylation is coordinately regulated by protein tyrosine kinases (PTKs) and protein tyrosine phosphatases 1 THE THREE-DIMENSIONAL STRUCTURE AND CATALYTIC (PTPs). Although PTKs add phosphate to tyrosine residues in MECHANISM OF PTPS proteins, PTPs remove it. Many PTKs are well-documented oncogenes.1 Recent cancer genomic studies provided compelling The three-dimensional structures of the catalytic domains of evidence that many PTPs function as tumor suppressor genes, classical PTPs (RPTPs and non-RPTPs) are extremely well because a majority of PTP mutations that have been identified in conserved.5 Even the catalytic domain structures of the dual- human cancers are loss-of-function mutations.