Molecular Mechanisms Underlying FIP1L1-PDGFRA– Mediated Myeloproliferation
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

FIP1L1 Gene FIP1 Like 1 (S
FIP1L1 gene FIP1 like 1 (S. cerevisiae) Normal Function The FIP1L1 gene provides instructions for making part of a protein complex named cleavage and polyadenylation specificity factor (CPSF). This complex of proteins plays an important role in processing molecules called messenger RNAs (mRNAs), which serve as the genetic blueprints for making proteins. The CPSF protein complex helps add a string of the RNA building block adenine to the mRNA, creating a polyadenine tail or poly(A) tail. The poly(A) tail is important for stability of the mRNA and for protein production from the blueprint. Health Conditions Related to Genetic Changes PDGFRA-associated chronic eosinophilic leukemia A deletion of genetic material from chromosome 4 brings together part of the FIP1L1 gene and part of another gene called PDGFRA, creating the FIP1L1-PDGFRA fusion gene. This mutation is a somatic mutation, which means it is acquired during a person's lifetime and is present only in certain cells. This fusion gene causes PDGFRA- associated chronic eosinophilic leukemia, which is a type of blood cell cancer characterized by an increased number of eosinophils, a type of white blood cell involved in allergic reactions. The FIP1L1-PDGFRA protein produced from the fusion gene has the function of the normal PDGFRA protein, which stimulates signaling pathways inside the cell that control many important cellular processes, such as cell growth and division (proliferation) and cell survival. Unlike the normal PDGFRA protein, however, the FIP1L1-PDGFRA protein is constantly turned on (constitutively activated), which means the cells are always receiving signals to proliferate. When the FIP1L1-PDGFRA fusion gene occurs in blood cell precursors, the growth of eosinophils (and occasionally other blood cells) is poorly controlled, leading to PDGFRA-associated chronic eosinophilic leukemia. -

R-Loop-Mediated Genome Instability in Mrna Cleavage and Polyadenylation Mutants
Downloaded from genesdev.cshlp.org on September 29, 2021 - Published by Cold Spring Harbor Laboratory Press R-loop-mediated genome instability in mRNA cleavage and polyadenylation mutants Peter C. Stirling,1,3 Yujia A. Chan,1,3 Sean W. Minaker,1,3 Maria J. Aristizabal,2 Irene Barrett,1 Payal Sipahimalani,1 Michael S. Kobor,2 and Philip Hieter1,4 1Michael Smith Laboratories, University of British Columbia, Vancouver, British Columbia V6T1Z4, Canada; 2Centre for Molecular Medicine and Therapeutics, Vancouver, British Columbia V5Z4H4, Canada Genome instability via RNA:DNA hybrid-mediated R loops has been observed in mutants involved in various aspects of transcription and RNA processing. The prevalence of this mechanism among essential chromosome instability (CIN) genes remains unclear. In a secondary screen for increased Rad52 foci in CIN mutants, representing ~25% of essential genes, we identified seven essential subunits of the mRNA cleavage and polyadenylation (mCP) machinery. Genome-wide analysis of fragile sites by chromatin immunoprecipitation (ChIP) and microarray (ChIP–chip) of phosphorylated H2A in these mutants supported a transcription-dependent mechanism of DNA damage characteristic of R loops. In parallel, we directly detected increased RNA:DNA hybrid formation in mCP mutants and demonstrated that CIN is suppressed by expression of the R-loop-degrading enzyme RNaseH. To investigate the conservation of CIN in mCP mutants, we focused on FIP1L1, the human ortholog of yeast FIP1, a conserved mCP component that is part of an oncogenic fusion in eosinophilic leukemia. We found that truncation fusions of yeast FIP1 analogous to those in cancer cause loss of function and that siRNA knockdown of FIP1L1 in human cells increases DNA damage and chromosome breakage. -

Hypereosinophilia in Granular Acute Bcell Lymphoblastic Leukemia With
bs_bs_banner Microscopic polyangiitis 543 and had been diagnosed with normocytic anemia.At that time, she References received treatment with an iron preparation because of a low 1 Ozen S, Ruperto N, Dillon MJ et al. EULAR/PReS endorsed con- serum iron level despite her normal ferritin level. We estimate that sensus criteria for the classification of childhood vasculitides. Ann. the patient already had mild alveolar hemorrhage at that time and Rheum. Dis. 2006; 65: 936–41. the bleeding later healed spontaneously. Among the cases of this 2 Vanoni F, Bettinelli A, Keller F, Bianchetti MG, Simonetti GD. disease reported from Japan, the disease tended to develop more Vasculitides associated with IgG antineutrophil cytoplasmic autoan- tibodies in childhood. Pediatr. Nephrol. 2010; 25: 205–12. frequently in the spring. In our patient, the disease developed twice 3 Kobayashi S, Inokuma S. Intrapulmonary hemorrhage in collagen- (both times in June), suggesting the possibility that infection vascular disease includes a spectrum of underlying conditions. triggered the disease onset. Although alveolar hemorrhage sub- Intern. Med. 2009; 48: 891–7. sided spontaneously in this case, lung fibrosis may develop in the 4 Hattori M, Kurayama H, Koitabashi Y. Antineutrophil cytoplasmic future if alveolar bleeding develops repeatedly and its diagnosis is autoantibody-associated glomerulonephritis in children. J. Am. Soc. Nephrol. 2001; 12: 1493–500. delayed. In the present case, treatment was started immediately 5 Peco-Antic A, Bonaci-Nikolic B, Basta-Jovanovic G et al. Child- after diagnosis and rapid aggravation of her renal function was hood microscopic polyangiitis associated with MPO-ANCA. prevented. When dealing with cases in whom unexplained nor- Pediatr. -

PRODUCT SPECIFICATION Prest Antigen FIP1L1 Product Datasheet
PrEST Antigen FIP1L1 Product Datasheet PrEST Antigen PRODUCT SPECIFICATION Product Name PrEST Antigen FIP1L1 Product Number APrEST79770 Gene Description factor interacting with PAPOLA and CPSF1 Alternative Gene DKFZp586K0717 Names Corresponding Anti-FIP1L1 (HPA037475) Antibodies Description Recombinant protein fragment of Human FIP1L1 Amino Acid Sequence Recombinant Protein Epitope Signature Tag (PrEST) antigen sequence: KANSSVGKWQDRYGRAESPDLRRLPGAIDVIGQTITISRVEGRRRANENS NIQVLSERSATEVDNNFSKPPPFFPPGAPPT Fusion Tag N-terminal His6ABP (ABP = Albumin Binding Protein derived from Streptococcal Protein G) Expression Host E. coli Purification IMAC purification Predicted MW 27 kDa including tags Usage Suitable as control in WB and preadsorption assays using indicated corresponding antibodies. Purity >80% by SDS-PAGE and Coomassie blue staining Buffer PBS and 1M Urea, pH 7.4. Unit Size 100 µl Concentration Lot dependent Storage Upon delivery store at -20°C. Avoid repeated freeze/thaw cycles. Notes Gently mix before use. Optimal concentrations and conditions for each application should be determined by the user. Product of Sweden. For research use only. Not intended for pharmaceutical development, diagnostic, therapeutic or any in vivo use. No products from Atlas Antibodies may be resold, modified for resale or used to manufacture commercial products without prior written approval from Atlas Antibodies AB. Warranty: The products supplied by Atlas Antibodies are warranted to meet stated product specifications and to conform to label descriptions when used and stored properly. Unless otherwise stated, this warranty is limited to one year from date of sales for products used, handled and stored according to Atlas Antibodies AB's instructions. Atlas Antibodies AB's sole liability is limited to replacement of the product or refund of the purchase price. -
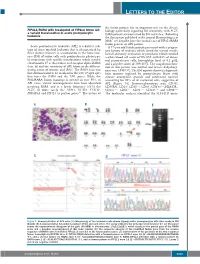
Letters to the Editor
LETTERS TO THE EDITOR the fusion partner has an important role on the disease FIP1L1/RARA with breakpoint at FIP1L1 intron 13: biology particularly regarding RA sensitivity, with PLZF- a variant translocation in acute promyelocytic 4 leukemia RARA patients characterized by RA resistance. Following the description published in the journal Haematologica in 2008, 5 we describe here the second case of FIP1L1/RARA fusion gene in an APL patient. Acute promyelocytic leukemia (APL) is a distinct sub - A 77-year old female patient presented with a progres - type of acute myeloid leukemia that is characterized by sive history of asthenia which lasted for several weeks. three distinct features: i) accumulation in the bone mar - Initial laboratory evaluation of peripheral blood revealed row (BM) of tumor cells with promyelocytic phenotype; a white blood cell count of 59 ¥10 9/L with 84% of abnor - ii) association with specific translocations which involve mal promyelocyte cells, hemoglobin level of 9.2 g/dL, chromosome 17 at the retinoic acid receptor alpha ( RARA ) and a platelet count of 109 ¥10 9/L. The coagulation func - locus ; iii) and the sensitivity of APL blasts to the differen - tion in this patient was normal and lactate dehydroge - 1 tiating action of retinoic acid (RA). The RARA locus was nase was 1,938 U/L. The BM aspirate showed a hypercel - first demonstrated to be involved in the t(15;17)(q22;q21) lular marrow replaced by promyelocyte blasts with that fuses the RARA and the PML genes. While the intense azurophilic granule and prominent nucleoli PML/RARA fusion transcript is present in over 95% of accounting for 93% of all nucleated cells, suggestive of APL cases, variant rearrangements have been identified APL (Figure 1A). -

ORIGINAL ARTICLE the Severity of FIP1L1–PDGFRA-Positive Chronic
Leukemia (2007) 21, 2428–2432 & 2007 Nature Publishing Group All rights reserved 0887-6924/07 $30.00 www.nature.com/leu ORIGINAL ARTICLE The severity of FIP1L1–PDGFRA-positive chronic eosinophilic leukaemia is associated with polymorphic variation at the IL5RA locus S Burgstaller1, S Kreil1, K Waghorn1, G Metzgeroth2, C Preudhomme3, K Zoi4, H White1, D Cilloni5, C Zoi4, F Brito-Babapulle6, C Walz2, A Reiter2 and NCP Cross1 1Wessex Regional Genetics Laboratory, University of Southampton, Salisbury, UK; 2III Medizinische Universita¨tsklinik, Medizinische Fakulta¨t der Universita¨t Mannheim, Heidelberg, Germany; 3Laboratoire d’Hematologie A, CHU Lille, Lille, France; 4Foundation of Biomedical Research, Academy of Athens, Athens, Greece; 5Department of Clinical and Biological Sciences, University of Turin, Turin, Italy and 6Department of Haematology, Ealing Hospital, London, UK We have investigated the hypothesis that constitutional genetic Understanding of the myeloproliferative subtype of HES was variation in IL-5 signalling may be associated with the greatly advanced by the finding of the FIP1L1–PDGFRA fusion, development or severity of FIP1L1–PDGFRA-positive chronic formed by a cytogenetically cryptic 800 kb interstitial deletion at eosinophilic leukaemia (CEL) in humans. We genotyped six 2 single-nucleotide polymorphisms (SNP) within or close to the chromosome band 4q12. Although initially described in more IL5RA or IL5 genes in 82 patients with FIP1L1–PDGFRA- than 50% of cases with idiopathic HES, subsequent studies positive CEL plus, as controls, healthy individuals (n ¼ 100), estimated the prevalence to be lower at 3À17%.3–5 Detection of patients with FIP1L1–PDGFRA-negative eosinophilia (n ¼ 100) FIP1L1–PDGFRA by reverse transcription-PCR or fluorescent in or patients with chronic myeloid leukaemia (CML) (n ¼ 100). -

FIP1L1‑PDGFRA Fusion‑Negative Hypereosinophilic Syndrome with Uncommon Cardiac Involvement Responding to Imatinib Treatment: a Case Report
MOLECULAR AND CLINICAL ONCOLOGY 9: 35-39, 2018 FIP1L1‑PDGFRA fusion‑negative hypereosinophilic syndrome with uncommon cardiac involvement responding to imatinib treatment: A case report AMANDA SANTOS DAL BERTO1, RICARDO HOHMANN CAMIÑA1, EDUARDO SILVA MACHADO1-3 and ANTUANI RAFAEL BAPTISTELLA1,2,4,5 1Santa Terezinha University Hospital; 2University of West Santa Catarina; 3Department of Clinical Oncology, Santa Terezinha University Hospital; 4Oncology Research Group of Santa Terezinha University Hospital /University of West Santa Catarina; 5Post Graduation Program in Bioscience and Health/University of West Santa Catarina, Joaçaba, Santa Catarina 89600-000, Brazil Received April 17, 2018; Accepted May 21, 2018 DOI: 10.3892/mco.2018.1637 Abstract. Hypereosinophilic syndrome is a rare, chronic therapeutic possibilities were evaluated due to the significant hematological disease characterized by a persistently elevated progression of the disease, and it was decided to attempt the eosinophil count exceeding 1.5x109/l, following the exclusion use of imatinib, despite its use being preferably recommended of other potential etiologies. The systemic involvement of the for FIPIL1-PDGFRA-positive patients. The patient exhibited an disease causes tissue damage through eosinophil infiltration, evident and immediate response to imatinib, with normalization and may affect various organs; cardiac complications are of the eosinophil count within 24 h of the first dose, which was observed in 50-60% of cases, which are predominately attrib- maintained for at least the next 19 months. This clinical presen- uted to endomyocardial fibrosis. The treatment is based initially tation is uncommon, as patients negative for FIPIL1-PDGFRA on determining the presence of the FIP1L1-PDGFRA fusion. fusion do not frequently respond to imatinib treatment, and Patients with positive results for this mutation tend to achieve symptomatic heart failure usually appears in the third stage of a complete response with imatinib treatment, which is thus disease progression. -

T-Cell Lymphoblastic Lymphoma Arising in the Setting of Myeloid/Lymphoid Neoplasms with Eosinophilia
cancers Article T-Cell Lymphoblastic Lymphoma Arising in the Setting of Myeloid/Lymphoid Neoplasms with Eosinophilia: LMO2 Immunohistochemistry as a Potentially Useful Diagnostic Marker Magda Zanelli 1 , Giuseppe G. Loscocco 2,3 , Elena Sabattini 4, Maurizio Zizzo 5,6,* , Francesca Sanguedolce 7, Luigi Panico 8, Daniela Fanni 9, Raffaella Santi 10, Cecilia Caprera 11, Cristiana Rossi 12, Alessandra Soriano 13,14, Alberto Cavazza 1, Alessandro Giunta 5, Cristina Mecucci 15, Alessandro M. Vannucchi 2,3, Stefano A. Pileri 16 and Stefano Ascani 11,15 1 Pathology Unit, Azienda Unità Sanitaria Locale—IRCCS di Reggio Emilia, 42123 Reggio Emilia, Italy; [email protected] (M.Z.); [email protected] (A.C.) 2 Department of Experimental and Clinical Medicine, University of Florence, 50134 Florence, Italy; gloscocco@unifi.it (G.G.L.); a.vannucchi@unifi.it (A.M.V.) 3 Center of Research and Innovation of Myeloproliferative Neoplasms (CRIMM), Azienda Ospedaliero-Universitaria Careggi, 50139 Florence, Italy 4 Citation: Zanelli, M.; Loscocco, G.G.; Haematopathology Unit, IRCCS Azienda Ospedaliero-Universitaria di Bologna, 40138 Bologna, Italy; Sabattini, E.; Zizzo, M.; [email protected] 5 Sanguedolce, F.; Panico, L.; Fanni, D.; Surgical Oncology Unit, Azienda Unità Sanitaria Locale—IRCCS di Reggio Emilia, 42123 Reggio Emilia, Italy; [email protected] Santi, R.; Caprera, C.; Rossi, C.; et al. 6 Clinical and Experimental Medicine PhD Program, University of Modena and Reggio Emilia, T-Cell Lymphoblastic Lymphoma 41121 Modena, Italy Arising in the Setting of 7 Pathology Unit, Azienda Ospedaliero-Universitaria—Ospedali Riuniti di Foggia, 71122 Foggia, Italy; Myeloid/Lymphoid Neoplasms with [email protected] Eosinophilia: LMO2 8 Pathology Unit Azienda Ospedaliera dei Colli Monaldi-Cotugno-CTO, P.O. -

Case Report Therapy-Related Acute Myeloid Leukemia with Eosinophilia, Basophilia, T(4;14)(Q12;Q24) and PDGFRA Rearrangement: A
Int J Clin Exp Pathol 2015;8(5):5812-5820 www.ijcep.com /ISSN:1936-2625/IJCEP0007951 Case Report Therapy-related acute myeloid leukemia with eosinophilia, basophilia, t(4;14)(q12;q24) and PDGFRA rearrangement: a case report and review of the literature Jun Zhou1, Peter Papenhausen2, Haipeng Shao1 1Department of Hematopathology and Laboratory Medicine, H. Lee Moffitt Cancer Center and Research Institute, 12902 Magnolia Drive, Tampa, FL, USA; 2Cytogenetics Laboratory, Laboratory Corporation of America, Research Triangle Park, NC, USA Received March 13, 2015; Accepted April 26, 2015; Epub May 1, 2015; Published May 15, 2015 Abstract: The myeloid and lymphoid neoplasms with eosinophilia and PDGFRA gene rearrangements usually show a good response to Imatinib and are typically associated with a normal karyotype, occasionally exhibiting a second- ary chromosomal abnormality associated with clonal evolution. Five variant translocations involving PDGFRA have been reported. Here, we report a rare case of therapy-related acute myeloid leukemia with PDGFRA rearrangement after chemotherapy for prior B lymphoblastic leukemia (B-ALL). The patient had a history of BCR-ABL negative, hy- podiploid B-ALL in complete remission after chemotherapy. However, 15 months later the patient developed acute myeloid leukemia with rapidly increasing eosinophilia, basophilia and a complex karyotype that included a novel t(4;14)(q12;q24). FIP1L1 was not associated with the PDGFRA rearrangement. The patient had a very aggressive clinical course, and died from the disease shortly after diagnosis. This is the first case of a primary therapy-related myeloid neoplasm with secondary PDGFRA rearrangement. The t(4:14)(q12;q24) is joining the growing list of the variant translocations involving PDGFRA. -

The Seventh Pathogenic Fusion Gene FIP1L1-RARA Was Isolated from a T
Letters to the Editor decade: life expectancy, long-term complication rates, and RARA gene. Following reverse transcription using total prognostic factors. Mayo Clin Proc 2006;81:159-66. mRNA from the patient’s bone marrow cells, the cDNA 12. De Stefano V, Za T, Rossi E, Vannucchi AM, Ruggeri M, obtained was ligated by T4 RNA ligase. The ligated prod- Elli E, et al. The GIMEMA CMD-Working Party. Recurrent thrombosis in patients with polycythemia vera and essen- uct was amplified by the nested polymerase chain reac- tial thrombocythemia: incidence, risk factors, and effect of tion (PCR). PCR primer sequences were as follows: 1st treatments. Haematologica 2008;93:372-80. PCR primers (5’-CTGCAGAAGTGCTTTGAAGT-3’, 5’- 13. Kittur J, Knudson RA, Lasho TL, Finke CM, Gangat N, CACCTTGTTGATGATGCAGT-3’) and 2nd PCR primers Wolanskyj AP, et al. Clinical correlates of JAK2V617F allele (5’-GAGTGCTCTGAGAGCTACAC-3’, 5’-CGGTGA- burden in essential thrombocythemia. Cancer 2007;109: 2279-84. CACGTGTACACCAT-3’). The products obtained were cloned and sequenced directly. As a result, FIP1L1 was identified as the fusion partner of RARA (Figure 1B). The The seventh pathogenic fusion gene FIP1L1-RARA RARA portion in this case starts, as expected, from exon was isolated from a t(4;17)-positive acute 3 and is fused to exon 15 of FIP1L1. While cloning the promyelocytic leukemia full length FIP1L1-RARA, we isolated three isoforms of FIP1L1-RARA; all of these isoforms are in-frame (Figure 1C). We also confirmed the mRNA expression of RARA- The majority of acute promyelocytic leukemia (APL) FIP1L1 by means of RT-PCR analysis (data not shown). -

Classic and Variants Apls, As Viewed from a Therapy Response
cancers Review Classic and Variants APLs, as Viewed from a Therapy Response Marie-Claude Geoffroy 1,2,3 and Hugues de Thé 1,2,3,4,5,* 1 Institut National de la Santé et de la Recherche Médicale (INSERM) U944, Equipe Labellisée par la Ligue Nationale contre le Cancer, 75010 Paris, France; marie-claude.geoff[email protected] 2 Centre National de la Recherche Scientifique Unité Mixte de Recherche 7212, Institut Universitaire d'Hématologie (IUH), 75010 Paris, France 3 Institut de Recherche Saint-Louis, Université de Paris, 75010 Paris, France 4 Assistance Publique-Hôpitaux de Paris, Service de Biochimie, Hôpital St-Louis, 75010 Paris, France 5 Collège de France, PSL Research University, INSERM U1050, CNRS UMR 7241, 75005 Paris, France * Correspondence: [email protected] Received: 24 March 2020; Accepted: 9 April 2020; Published: 14 April 2020 Abstract: Most acute promyelocytic leukemia (APL) are caused by PML-RARA, a translocation-driven fusion oncoprotein discovered three decades ago. Over the years, several other types of rare X-RARA fusions have been described, while recently, oncogenic fusion proteins involving other retinoic acid receptors (RARB or RARG) have been associated to very rare cases of acute promyelocytic leukemia. PML-RARA driven pathogenesis and the molecular basis for therapy response have been the focus of many studies, which have now converged into an integrated physio-pathological model. The latter is well supported by clinical and molecular studies on patients, making APL one of the rare hematological disorder cured by targeted therapies. Here we review recent data on APL-like diseases not driven by the PML-RARA fusion and discuss these in view of current understanding of “classic” APL pathogenesis and therapy response. -

Diagnosis and Treatment of Myeloproliferative Neoplasms with Eosinophilia
Myeloproliferative neoplasms Diagnosis and treatment of myeloproliferative neoplasms with eosinophilia A. Reiter 1 ABSTRACT G. Metzgeroth 1 Significant eosinophilia is a rare but recurrent morphological feature of all myeloid neoplasms but N.C.P. Cross 2,3 the correct diagnosis of eosinophilia-associated disorders remains problematic. The WHO 2008 clas - sification defines a rare subgroup characterized by selected tyrosine kinase (TK) fusion genes: ‘myeloid and lymphoid neoplasms with eosinophilia and abnormalities of PDGFRA, PDGFRB or FGFR1 ’ (MLN-eo), 1 III. Medizinische Klinik, of which by far the most common fusion gene is FIP1L1-PDGFRA . For other cases, proof of clonality Universitätsmedizin Mannheim, by the finding of increased numbers of blasts and/or cytogenetic or molecular aberrations is the basis Mannheim, Germany 2 for a diagnosis of chronic eosinophilic leukemia, not otherwise specified (CEL-NOS), however this is Wessex Regional Genetics only possible infrequently. To date, over 50 fusion genes encoding deregulated TK have been identified Laboratory, Salisbury, UK 3Faculty of Medicine, University of within the broad spectrum of eosinophilia-associated myeloproliferative neoplasms (MPN-eo) or Southampton, Southampton, UK myelodysplastic/myeloproliferative neoplasms (MDS/MPN-eo). Important to recognize are also those cases that do not fulfil the diagnostic criteria for MLN-eo or CEL-NOS but who may harbor TK point mutations, e.g. KIT D816V positive systemic mastocytosis. For treatment decisions, disease stage Correspondence: (chronic/blast phase), potential clinical course (indolent/aggressive), sensitivity to imatinib or alterna - Andreas Reiter E-mail: andreas.reiter@medma. tive TK inhibitors ( de novo /resistant disease) and eligibility for allogeneic stem cell transplantation uni-heidelberg.de need to be taken into account on an individual basis.