Mitophagy Modulators
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-
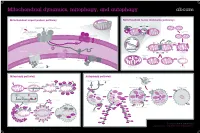
Mitochondrial Dynamics, Mitophagy, and Autophagy
Mitochondrial dynamics, mitophagy, and autophagy Mitochondrial import protein pathway Mitochondrial fusion and fission pathways iΔΨm, (OXPHOSh) Ca2+ Fusion ++++ (Cell differentiationi) N Internal signal β-barrel outer Opa1 SH ER sequence SH membrane precursors Mfn1/2 SH SH GTP GTP C Inner membrane and GTP GTP Outer membrane Matrix carrier precursors 22 70 GTP fusion (N-terminal presequences) 35 GTP 20 37 Tom complex 5 Sam complex Opa1 Cytosol Outer 6 Sam50 7 Mdm10 membrane DRP1 Fis1 Inner membrane fusion Tim8-Tim13 Mim 1 Tom40 Bax/Bak apoptosis Outer membrane HR2 regions S-S Tim9-Tim10 S-S Inner Erv1 95 A 54 mitochondrial S-S Reorganization Mia40 space 21 Fission sequestration 50 18 Tim22 Translocation Depolarization to meet iΔΨm ATP needs Tim23 KREBS Cycle NADH FADH2 17 Oxa1 Tim 22 complex Mia Mba1 Mdm I ATP 44 38 H+ II 17 H+ Inner membrane 16 +++ III ADP - Pi 18 Ribosome Oxa H+ IV mtHsp70 H+ ATP +++ Mge1 MPP Healthy mitochondrion Mitophagy Matrix Tim 23 complex Mitophagy pathway Autophagy pathway Lysosomal hydroiase Lysosome Hypoxia, Nutrient/Growth Rapamycin Starvation condition Ubiquitin Cytosol Parkin factor deprivation Lamp Stress Damaged mitochondrion Parkin Parkin iΔΨm Parkin MARF PINK1 PINK1 PINK1 Mfn1 VDAC1 Parkin AMPK mTOR Mfn2 PINK1 PINK1 Phagophore Autophagosome Autophagolyosome PARL LC3-II PINK1-L PINK1-S Parkin P FIS1 ATG13 ULK LC3-II Membrane PINK1 PINK1 FIP200 PINK1 MIRO2 P Bak PINK1 MIRO1 Parkin Parkin Parkin Cytoplasmic Cytosol proLC3 macromolecule LC3-II Atg4B Atg7/LC3-I Atg5/Atg12/Atg16 Organelle Atg9 PE Atg18 Atg2 -

Involvement of Impaired Autophagy and Mitophagy in Neuro-2A Cell
www.nature.com/scientificreports OPEN Involvement of impaired autophagy and mitophagy in Neuro-2a cell damage under hypoxic and/or high- Received: 19 May 2017 Accepted: 15 January 2018 glucose conditions Published: xx xx xxxx Yufei Song, Yu Du, Wenying Zou, Yan Luo, Xiaojie Zhang & Jianliang Fu Chronic cerebral hypoperfusion (CCH) plays an insidious role in the development of cognitive impairment. Considerable evidence suggests that Diabetes Mellitus (DM) as a vascular risk factor may exacerbate CCH and is closely related to cognitive decline. Dysregulation of autophagy is known to be associated with the pathogenesis of neurodegenerative diseases such as Alzheimer’s disease. To elucidate the role of autophagy in CCH- and/or DM-related pathogenesis, mouse neuroblastoma Neuro-2a cells were exposed to hypoxia and/or high glucose for 48 h, mimicking CCH complicated with DM pathologies. Chronic hypoxia reduced cell proliferation and increased levels of cleaved caspase-3, whereas high glucose had no obvious synergistic toxic efect. Accumulation of autophagic vacuoles under hypoxia may be due to both autophagy impairment and induction, with the former accounting for Neuro-2a cell death. Additionally, aberrant accumulation of mitochondria in Neuro-2a cells may be attributed to insufcient BNIP3-mediated mitophagy due to poor interaction between BNIP3 and LC3-II. Despite the lack of a signifcant cytotoxic efect of high glucose under our experimental conditions, our data indicated for the frst time that impaired autophagy degradation and inefcient BNIP3-mediated mitophagy may constitute mechanisms underlying neuronal cell damage during chronic hypoxia. Chronic cerebral hypoperfusion (CCH) is a normal process related to ageing that likely contributes to age-related memory loss1. -

Compression of Large Sets of Sequence Data Reveals Fine Diversification of Functional Profiles in Multigene Families of Proteins
Technical note Compression of Large Sets of Sequence Data Reveals Fine Diversification of Functional Profiles in Multigene Families of Proteins: A Study for Peptidyl-Prolyl cis/trans Isomerases (PPIase) Andrzej Galat Retired from: Service d’Ingénierie Moléculaire des Protéines (SIMOPRO), CEA-Université Paris-Saclay, France; [email protected]; Tel.: +33-0164465072 Received: 21 December 2018; Accepted: 21 January 2019; Published: 11 February 2019 Abstract: In this technical note, we describe analyses of more than 15,000 sequences of FK506- binding proteins (FKBP) and cyclophilins, also known as peptidyl-prolyl cis/trans isomerases (PPIases). We have developed a novel way of displaying relative changes of amino acid (AA)- residues at a given sequence position by using heat-maps. This type of representation allows simultaneous estimation of conservation level in a given sequence position in the entire group of functionally-related paralogues (multigene family of proteins). We have also proposed that at least two FKBPs, namely FKBP36, encoded by the Fkbp6 gene and FKBP51, encoded by the Fkbp5 gene, can form dimers bound via a disulfide bridge in the nucleus. This type of dimer may have some crucial function in the regulation of some nuclear complexes at different stages of the cell cycle. Keywords: FKBP; cyclophilin; PPIase; heat-map; immunophilin 1 Introduction About 30 years ago, an exciting adventure began in finding some correlations between pharmacological activities of macrocyclic hydrophobic drugs, namely the cyclic peptide cyclosporine A (CsA), and two macrolides, namely FK506 and rapamycin, which have profound and clinically useful immunosuppressive effects, especially in organ transplantations and in combating some immune disorders. -

FKBP8 Antibody Cat
FKBP8 Antibody Cat. No.: 62-126 FKBP8 Antibody With HepG2 cell line lysate, the resolved proteins were electrophoretically transferred to PVDF membrane and incubated sequentially with primary antibody FKBP38 (1:1000, 4˚C,overnight ) and horseradish peroxidase–conjugated second antibody. After washing, the bound antibody complex was detected using an ECL chemiluminescence reagentand XAR film (Kodak). Specifications HOST SPECIES: Rabbit SPECIES REACTIVITY: Human HOMOLOGY: Predicted species reactivity based on immunogen sequence: Mouse, Rat This FKBP8 antibody is generated from rabbits immunized with a KLH conjugated IMMUNOGEN: synthetic peptide between 199-226 amino acids from the Central region of human FKBP8. TESTED APPLICATIONS: WB APPLICATIONS: For WB starting dilution is: 1:1000 PREDICTED MOLECULAR 45 kDa WEIGHT: September 28, 2021 1 https://www.prosci-inc.com/fkbp8-antibody-62-126.html Properties This antibody is purified through a protein A column, followed by peptide affinity PURIFICATION: purification. CLONALITY: Polyclonal ISOTYPE: Rabbit Ig CONJUGATE: Unconjugated PHYSICAL STATE: Liquid BUFFER: Supplied in PBS with 0.09% (W/V) sodium azide. CONCENTRATION: batch dependent Store at 4˚C for three months and -20˚C, stable for up to one year. As with all antibodies STORAGE CONDITIONS: care should be taken to avoid repeated freeze thaw cycles. Antibodies should not be exposed to prolonged high temperatures. Additional Info OFFICIAL SYMBOL: FKBP8 Peptidyl-prolyl cis-trans isomerase FKBP8, PPIase FKBP8, 38 kDa FK506-binding protein, ALTERNATE NAMES: 38 kDa FKBP, FKBP-38, hFKBP38, FK506-binding protein 8, FKBP-8, FKBPR38, Rotamase, FKBP8, FKBP38 ACCESSION NO.: Q14318 PROTEIN GI NO.: 193806337 GENE ID: 23770 USER NOTE: Optimal dilutions for each application to be determined by the researcher. -

Naringenin Regulates FKBP4/NR3C1/TMEM173 Signaling Pathway in Autophagy and Proliferation of Breast Cancer and Tumor-Infltrating Dendritic Cell Maturation
Naringenin Regulates FKBP4/NR3C1/TMEM173 Signaling Pathway in Autophagy and Proliferation of Breast Cancer and Tumor-Inltrating Dendritic Cell Maturation Hanchu Xiong ( [email protected] ) Zhejiang Provincial People's Hospital https://orcid.org/0000-0001-6075-6895 Zihan Chen First Hospital of Zhejiang Province: Zhejiang University School of Medicine First Aliated Hospital Baihua Lin Zhejiang Provincial People's Hospital Cong Chen Zhejiang University School of Medicine Sir Run Run Shaw Hospital Zhaoqing Li Zhejiang University School of Medicine Sir Run Run Shaw Hospital Yongshi Jia Zhejiang Provincial People's Hospital Linbo Wang Zhejiang University School of Medicine Sir Run Run Shaw Hospital Jichun Zhou Zhejiang University School of Medicine Sir Run Run Shaw Hospital Research Keywords: FKBP4, TMEM173, Autophagy, Exosome, Dendritic cell, Breast cancer Posted Date: July 7th, 2021 DOI: https://doi.org/10.21203/rs.3.rs-659646/v1 License: This work is licensed under a Creative Commons Attribution 4.0 International License. Read Full License Page 1/38 Abstract Background TMEM173 is a pattern recognition receptor detecting cytoplasmic nucleic acids and transmits cGAS related signals that activate host innate immune responses. It has also been found to be involved in tumor immunity and tumorigenesis. Methods Bc-GenExMiner, PROMO and STRING database were used for analyzing clinical features and interplays of FKBP4, TMEM173 and NR3C1. Transient transfection, western blotting, quantitative real-time PCR, luciferase reporter assay, immunouorescence and nuclear and cytoplasmic fractionation were used for regulation of FKBP4, TMEM173 and NR3C1. Both knockdown and overexpression of FKBP4, TMEM173 and NR3C1 were used to analyze effects on autophagy and proliferation of breast cancer (BC) cells. -

SI Appendix Table of Contents
SI Appendix Contact-ID, a tool for profiling organelle-membrane contact sites, reveals regulatory proteins of mitochondrial-associated membrane formation Chulhwan Kwak1,2†, Sanghee Shin3,4,†, Jong-Seok Park2,†, Minkyo Jung5, Truong Thi My Nhung6, Myeong-Gyun Kang1,2, Chaiheon Lee2, Tae-Hyuk Kwon2, Sang Ki Park6,*, Ji Young Mun5,*, Jong-Seo Kim3,4,*, and Hyun-Woo Rhee1,4* 1Department of Chemistry, Seoul National University, Seoul, 08826, Korea 2Department of Chemistry, Ulsan National Institute of Science and Technology (UNIST), Ulsan, 44919, Korea 3Center for RNA Research, Institute of Basic Science (IBS), Seoul, 08826, Korea 4School of Biological Sciences, Seoul National University, Seoul, 08826, Korea 5Department of Structure and Function of Neural Network, Korea Brain Research Institute, Daegu, 41062, South Korea 6 Department of Life Sciences, Pohang University of Science and Technology (POSTECH), 37673, Pohang, Republic of Korea. *Corresponding authors: [email protected] (H.-W.R.), [email protected] (J.-S.K.), [email protected] (J. Y. M.), [email protected] (S. K. P.) † These authors equally contributed to this work. Table of Contents Table S1------------------------------------------------------------------------------------------------------------------------------------------------S3 Figure S1-17------------------------------------------------------------------------------------------------------------------------------S4-32 Movie S1-4& dataset S1-5 legends-------------------------------------------------------------------------------------------------------S33 -

Mitoq Inhibits Hepatic Stellate Cell Activation and Liver Fibrosis by Enhancing PINK1/Parkin-Mediated Mitophagy
MitoQ Inhibits Hepatic Stellate Cell Activation and Liver Fibrosis by Enhancing PINK1/parkin-mediated Mitophagy Shi-Ying Dou Second Hospital of Hebei Medical University Jiu-Na Zhang Second Hospital of Hebei Medical University Xiao-Li Xie Second Hospital of Hebei Medical University Ting Liu Second Hospital of Hebei Medical University Jun-Li Hu Second Hospital of Hebei Medical University Xiao-Yu Jiang Second Hospital of Hebei Medical University Miao-Miao Wang Second Hospital of Hebei Medical University Hui-Qing Jiang ( [email protected] ) Second Hospital of Hebei Medical University https://orcid.org/0000-0002-5235-1457 Research Article Keywords: Liver brosis, hepatic stellate cell, ubiquinone, PINK1 mitophagy Posted Date: March 30th, 2021 DOI: https://doi.org/10.21203/rs.3.rs-344963/v1 License: This work is licensed under a Creative Commons Attribution 4.0 International License. Read Full License Page 1/18 Abstract Mitophagy plays an important role in the activation of hepatic stellate cells (HSCs). Mitochondria- targeted ubiquinone (MitoQ) is a mitochondria-targeted antioxidant that reduces the production of intracellular reactive oxygen species (ROS). However, its relationship with mitophagy remains unclear. This study evaluated mitophagy during HSC activation and the effects of MitoQ on mitophagy in cell culture and in an animal model of the activation of HSCs. We found that MitoQ reduced the activation of HSCs and alleviated hepatic brosis. While activation of primary HSCs or LX-2 cells was associated with reduced PINK1/parkin-mediated mitophagy, MitoQ reduced intracellular ROS levels, enhanced PINK1/parkin-mediated mitophagy, and inhibited the activation of HSCs. After knocking down the key mitophagy-related protein, PINK1, in LX-2 cells to block mitophagy, MitoQ intervention failed to inhibit HSC activation. -

ZMYND10 Functions in a Chaperone Relay During Axonemal Dynein
RESEARCH ARTICLE ZMYND10 functions in a chaperone relay during axonemal dynein assembly Girish R Mali1†‡, Patricia L Yeyati1†, Seiya Mizuno2, Daniel O Dodd1, Peter A Tennant1, Margaret A Keighren1, Petra zur Lage3, Amelia Shoemark4, Amaya Garcia-Munoz5, Atsuko Shimada6, Hiroyuki Takeda6, Frank Edlich7,8, Satoru Takahashi2,9, Alex von Kreigsheim5,10, Andrew P Jarman3, Pleasantine Mill1* 1MRC Human Genetics Unit, Institute of Genetics and Molecular Medicine, University of Edinburgh, Edinburgh, United Kingdom; 2Laboratory Animal Resource Centre, University of Tsukuba, Tsukuba, Japan; 3Centre for Discovery Brain Sciences, University of Edinburgh, Edinburgh, United Kingdom; 4Division of Molecular and Clinical Medicine, University of Dundee, Dundee, United Kingdom; 5Systems Biology Ireland, University College Dublin, Dublin, Ireland; 6Department of Biological Sciences, University of Tokyo, Tokyo, Japan; 7Institute for Biochemistry and Molecular Biology, University of Freiburg, Freiburg, Germany; 8BIOSS, Centre for Biological Signaling Studies, University of Freiburg, Freiburg, Germany; 9Department of Anatomy and Embryology, Faculty of Medicine, University of Tsukuba, Tsukuba, Japan; 10Edinburgh Cancer Research UK Centre, Institute of Genetics and Molecular Medicine, University of Edinburgh, Edinburgh, United Kingdom *For correspondence: [email protected] †These authors contributed Abstract Molecular chaperones promote the folding and macromolecular assembly of a diverse equally to this work set of ‘client’ proteins. How ubiquitous chaperone machineries direct their activities towards specific sets of substrates is unclear. Through the use of mouse genetics, imaging and quantitative Present address: ‡MRC Laboratory of Molecular Biology, proteomics we uncover that ZMYND10 is a novel co-chaperone that confers specificity for the Cambridge, United Kingdom FKBP8-HSP90 chaperone complex towards axonemal dynein clients required for cilia motility. -

Gp78 E3 Ubiquitin Ligase Mediates Both Basal and Damage-Induced Mitophagy
bioRxiv preprint doi: https://doi.org/10.1101/407593; this version posted September 3, 2018. The copyright holder for this preprint (which was not certified by peer review) is the author/funder. All rights reserved. No reuse allowed without permission. Gp78 E3 ubiquitin ligase mediates both basal and damage-induced mitophagy Bharat Joshi, Yahya Mohammadzadeh, Guang Gao and Ivan R. Nabi Department of Cellular and Physiological Sciences, Life Sciences Institute, University of British Columbia, Vancouver, BC V6T 1Z3, Canada #Running title: Gp78 control of mitophagy §To whom correspondence should be addressed: Ivan R. Nabi, Department of Cellular and Physiological Sciences, Life Sciences Institute, University of British Columbia, 2350 Health Sciences Mall, Vancouver, BC V6T 1Z3 Canada. Tel: +1-(604) 822-7000 E-mail: [email protected] Key words: Gp78 ubiquitin ligase; mitochondria; autophagy; PINK1; Parkin bioRxiv preprint doi: https://doi.org/10.1101/407593; this version posted September 3, 2018. The copyright holder for this preprint (which was not certified by peer review) is the author/funder. All rights reserved. No reuse allowed without permission. Abstract Mitophagy, the elimination of mitochondria by the autophagy machinery, evolved to monitor mitochondrial health and maintain mitochondrial integrity. PINK1 is a sensor of mitochondrial health that recruits Parkin and other mitophagy-inducing ubiquitin ligases to depolarized mitochondria. However, mechanisms underlying mitophagic control of mitochondrial homeostasis, basal mitophagy, remain poorly understood. The Gp78 E3 ubiquitin ligase, an endoplasmic reticulum membrane protein, induces mitochondrial fission, endoplasmic reticulum- mitochondria contacts and mitophagy of depolarized mitochondria. CRISPR/Cas9 knockout of Gp78 in HT-1080 fibrosarcoma cells results in reduced ER-mitochondria contacts, increased mitochondrial volume and resistance to CCCP-induced mitophagy. -

Mitochondrial Physiology Within Myelinated Axons in Health and Disease : an Energetic Interplay Between Counterparts Gerben Van Hameren
Mitochondrial physiology within myelinated axons in health and disease : an energetic interplay between counterparts Gerben Van Hameren To cite this version: Gerben Van Hameren. Mitochondrial physiology within myelinated axons in health and disease : an energetic interplay between counterparts. Human health and pathology. Université Montpellier, 2018. English. NNT : 2018MONTT084. tel-02053421 HAL Id: tel-02053421 https://tel.archives-ouvertes.fr/tel-02053421 Submitted on 1 Mar 2019 HAL is a multi-disciplinary open access L’archive ouverte pluridisciplinaire HAL, est archive for the deposit and dissemination of sci- destinée au dépôt et à la diffusion de documents entific research documents, whether they are pub- scientifiques de niveau recherche, publiés ou non, lished or not. The documents may come from émanant des établissements d’enseignement et de teaching and research institutions in France or recherche français ou étrangers, des laboratoires abroad, or from public or private research centers. publics ou privés. THÈSE POUR OBTENIR LE GRADE DE DOCTEUR DE L’UNIVERSITÉ DE M ONTPELLIER En Biologie Santé École doctorale CBS2 Institut des Neurosciences de Montpellier MITOCHONDRIAL PHYSIOLOGY WITHIN MYELINATED AXONS IN HEALTH AND DISEASE AN ENERGETIC INTERPLAY BETWEEN COUNTERPARTS Présentée par Gerben van Hameren Le 23 Novembre 2018 Sous la direction de Dr. Nicolas Tricaud Devant le jury composé de Prof. Pascale Belenguer, Centre de Recherches sur la Cognition Animale Toulouse Professeur d’université Dr. Guy Lenaers, Mitochondrial Medicine Research Centre Angers Directeur de recherche Dr. Don Mahad, University of Edinburgh Senior clinical lecturer Invité Dr. Marie-Luce Vignais, Institute for Regenerative Medicine & Biotherapy, Montpellier Chargé de recherche 0 Table of contents Prologue ................................................................................................................................................. -

In Vivo Real-Time Dynamics of ATP and ROS Production in Axonal
Hameren et al. Acta Neuropathologica Communications (2019) 7:86 https://doi.org/10.1186/s40478-019-0740-4 RESEARCH Open Access In vivo real-time dynamics of ATP and ROS production in axonal mitochondria show decoupling in mouse models of peripheral neuropathies Gerben van Hameren1* , Graham Campbell1, Marie Deck1, Jade Berthelot1, Benoit Gautier1, Patrice Quintana1, Roman Chrast2,3 and Nicolas Tricaud1* Abstract Mitochondria are critical for the function and maintenance of myelinated axons notably through Adenosine triphosphate (ATP) production. A direct by-product of this ATP production is reactive oxygen species (ROS), which are highly deleterious for neurons. While ATP shortage and ROS levels increase are involved in several neurodegenerative diseases, it is still unclear whether the real-time dynamics of both ATP and ROS production in axonal mitochondria are altered by axonal or demyelinating neuropathies. To answer this question, we imaged and quantified mitochondrial ATP and hydrogen peroxide (H2O2) in resting or stimulated peripheral nerve myelinated axons in vivo, using genetically-encoded fluorescent probes, two-photon time-lapse and CARS imaging. We found that ATP and H2O2 productions are intrinsically higher in nodes of Ranvier even in resting conditions. Axonal firing increased both ATP and H2O2 productions but with different dynamics: ROS production peaked shortly and transiently after the stimulation while ATP production increased gradually for a longer period of time. In neuropathic MFN2R94Q mice, mimicking Charcot-Marie-Tooth 2A disease, defective mitochondria failed to upregulate ATP production following axonal activity. However, elevated H2O2 production was largely sustained. Finally, inducing demyelination with lysophosphatidylcholine resulted in a reduced level of ATP while H2O2 level soared. -

Roles of Sigma-1 Receptors on Mitochondrial Functions Relevant to Neurodegenerative Diseases Tzu-Yu Weng1,2, Shang-Yi Anne Tsai1 and Tsung-Ping Su1*
Weng et al. Journal of Biomedical Science (2017) 24:74 DOI 10.1186/s12929-017-0380-6 REVIEW Open Access Roles of sigma-1 receptors on mitochondrial functions relevant to neurodegenerative diseases Tzu-Yu Weng1,2, Shang-Yi Anne Tsai1 and Tsung-Ping Su1* Abstract The sigma-1 receptor (Sig-1R) is a chaperone that resides mainly at the mitochondrion-associated endoplasmic reticulum (ER) membrane (called the MAMs) and acts as a dynamic pluripotent modulator in living systems. At the MAM, the Sig-1R is known to play a role in regulating the Ca2+ signaling between ER and mitochondria and in maintaining the structural integrity of the MAM. The MAM serves as bridges between ER and mitochondria regulating multiple functions such as Ca2+ transfer, energy exchange, lipid synthesis and transports, and protein folding that are pivotal to cell survival and defense. Recently, emerging evidences indicate that the MAM is critical in maintaining neuronal homeostasis. Thus, given the specific localization of the Sig-1R at the MAM, we highlight and propose that the direct or indirect regulations of the Sig-1R on mitochondrial functions may relate to neurodegenerative diseases including Alzheimer’s disease (AD), Parkinson’s disease (PD), Huntington’s disease (HD) and amyotrophic lateral sclerosis (ALS). In addition, the promising use of Sig-1R ligands to rescue mitochondrial dysfunction-induced neurodegeneration is addressed. Keywords: Sigma-1 receptor, Mitochondria, Mitochondrion-associated ER membrane (MAM), Neurodegenerative disorders Background also regulates Ca2+ influx by attenuating the coupling of The sigma-1 receptor (Sig-1R) is an endoplasmic the ER Ca2+ sensor STIM1 to Orai1 [3].