Ultra-Fast and Accurate Motif Finding in Large Chip-Seq Datasets Reveals Transcription Factor Binding Patterns
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Transcription Factor P73 Regulates Th1 Differentiation
ARTICLE https://doi.org/10.1038/s41467-020-15172-5 OPEN Transcription factor p73 regulates Th1 differentiation Min Ren1, Majid Kazemian 1,4, Ming Zheng2, JianPing He3, Peng Li1, Jangsuk Oh1, Wei Liao1, Jessica Li1, ✉ Jonathan Rajaseelan1, Brian L. Kelsall 3, Gary Peltz 2 & Warren J. Leonard1 Inter-individual differences in T helper (Th) cell responses affect susceptibility to infectious, allergic and autoimmune diseases. To identify factors contributing to these response differ- 1234567890():,; ences, here we analyze in vitro differentiated Th1 cells from 16 inbred mouse strains. Haplotype-based computational genetic analysis indicates that the p53 family protein, p73, affects Th1 differentiation. In cells differentiated under Th1 conditions in vitro, p73 negatively regulates IFNγ production. p73 binds within, or upstream of, and modulates the expression of Th1 differentiation-related genes such as Ifng and Il12rb2. Furthermore, in mouse experimental autoimmune encephalitis, p73-deficient mice have increased IFNγ production and less dis- ease severity, whereas in an adoptive transfer model of inflammatory bowel disease, transfer of p73-deficient naïve CD4+ T cells increases Th1 responses and augments disease severity. Our results thus identify p73 as a negative regulator of the Th1 immune response, suggesting that p73 dysregulation may contribute to susceptibility to autoimmune disease. 1 Laboratory of Molecular Immunology and the Immunology Center, National Heart, Lung, and Blood Institute, Bethesda, MD 20892-1674, USA. 2 Department of Anesthesia, Stanford University School of Medicine, Stanford, CA 94305, USA. 3 Laboratory of Molecular Immunology, National Institute of Allergy and Infectious Diseases, Bethesda, MD 20892, USA. 4Present address: Department of Biochemistry and Computer Science, Purdue University, West ✉ Lafayette, IN 37906, USA. -
![Overlap of Vitamin a and Vitamin D Target Genes with CAKUT- Related Processes [Version 1; Peer Review: 1 Approved with Reservations]](https://docslib.b-cdn.net/cover/4295/overlap-of-vitamin-a-and-vitamin-d-target-genes-with-cakut-related-processes-version-1-peer-review-1-approved-with-reservations-144295.webp)
Overlap of Vitamin a and Vitamin D Target Genes with CAKUT- Related Processes [Version 1; Peer Review: 1 Approved with Reservations]
F1000Research 2021, 10:395 Last updated: 21 JUL 2021 BRIEF REPORT Overlap of vitamin A and vitamin D target genes with CAKUT- related processes [version 1; peer review: 1 approved with reservations] Ozan Ozisik1, Friederike Ehrhart 2,3, Chris T Evelo 2, Alberto Mantovani4, Anaı̈s Baudot 1,5 1Aix Marseille University, Inserm, MMG, Marseille, 13385, France 2Department of Bioinformatics - BiGCaT, Maastricht University, Maastricht, 6200 MD, The Netherlands 3Department of Bioinformatics, NUTRIM/MHeNs, Maastricht University, Maastricht, 6200 MD, The Netherlands 4Istituto Superiore di Sanità, Rome, 00161, Italy 5Barcelona Supercomputing Center (BSC), Barcelona, 08034, Spain v1 First published: 18 May 2021, 10:395 Open Peer Review https://doi.org/10.12688/f1000research.51018.1 Latest published: 18 May 2021, 10:395 https://doi.org/10.12688/f1000research.51018.1 Reviewer Status Invited Reviewers Abstract Congenital Anomalies of the Kidney and Urinary Tract (CAKUT) are a 1 group of abnormalities affecting the kidneys and their outflow tracts, which include the ureters, the bladder, and the urethra. CAKUT version 1 patients display a large clinical variability as well as a complex 18 May 2021 report aetiology, as only 5% to 20% of the cases have a monogenic origin. It is thereby suspected that interactions of both genetic and 1. Elena Menegola, Università degli Studi di environmental factors contribute to the disease. Vitamins are among the environmental factors that are considered for CAKUT aetiology. In Milano, Milan, Italy this study, we collected vitamin A and vitamin D target genes and Any reports and responses or comments on the computed their overlap with CAKUT-related gene sets. -

Transcriptomic Characterization of Fibrolamellar Hepatocellular
Transcriptomic characterization of fibrolamellar PNAS PLUS hepatocellular carcinoma Elana P. Simona, Catherine A. Freijeb, Benjamin A. Farbera,c, Gadi Lalazara, David G. Darcya,c, Joshua N. Honeymana,c, Rachel Chiaroni-Clarkea, Brian D. Dilld, Henrik Molinad, Umesh K. Bhanote, Michael P. La Quagliac, Brad R. Rosenbergb,f, and Sanford M. Simona,1 aLaboratory of Cellular Biophysics, The Rockefeller University, New York, NY 10065; bPresidential Fellows Laboratory, The Rockefeller University, New York, NY 10065; cDivision of Pediatric Surgery, Department of Surgery, Memorial Sloan-Kettering Cancer Center, New York, NY 10065; dProteomics Resource Center, The Rockefeller University, New York, NY 10065; ePathology Core Facility, Memorial Sloan-Kettering Cancer Center, New York, NY 10065; and fJohn C. Whitehead Presidential Fellows Program, The Rockefeller University, New York, NY 10065 Edited by Susan S. Taylor, University of California, San Diego, La Jolla, CA, and approved September 22, 2015 (received for review December 29, 2014) Fibrolamellar hepatocellular carcinoma (FLHCC) tumors all carry a exon of DNAJB1 and all but the first exon of PRKACA. This deletion of ∼400 kb in chromosome 19, resulting in a fusion of the produced a chimeric RNA transcript and a translated chimeric genes for the heat shock protein, DNAJ (Hsp40) homolog, subfam- protein that retains the full catalytic activity of wild-type PKA. ily B, member 1, DNAJB1, and the catalytic subunit of protein ki- This chimeric protein was found in 15 of 15 FLHCC patients nase A, PRKACA. The resulting chimeric transcript produces a (21) in the absence of any other recurrent mutations in the DNA fusion protein that retains kinase activity. -

Roles of Id3 and IL-13 in a Mouse Model of Autoimmune Exocrinopathy
Roles of Id3 and IL-13 in a Mouse Model of Autoimmune Exocrinopathy by Ian Lawrence Belle Department of Immunology Duke University Date:_______________________ Approved: ___________________________ Yuan Zhuang, Supervisor ___________________________ Michael Krangel, Chair ___________________________ Qi-jing Li ___________________________ Richard Lee Reinhardt ___________________________ Arno Greenleaf Dissertation submitted in partial fulfillment of the requirements for the degree of Doctor of Philosophy in the Department of Immunology in the Graduate School of Duke University 2015 ABSTRACT Roles of Id3 and IL-13 in a Mouse Model of Autoimmune Exocrinopathy by Ian Lawrence Belle Department of Immunology Duke University Date:_______________________ Approved: ___________________________ Yuan Zhuang, Supervisor ___________________________ Michael Krangel, Chair ___________________________ Qi-jing Li ___________________________ Richard Lee Reinhardt ___________________________ Arno Greenleaf An abstract of a dissertation submitted in partial fulfillment of the requirements for the degree of Doctor of Philosophy in the Department of Immunology in the Graduate School of Duke University 2015 Copyright by Ian Lawrence Belle 2015 Abstract Within the field of immunology, the existence of autoimmune diseases presents a unique set of challenges. The immune system typically protects the host by identifying foreign pathogens and mounting an appropriate response to eliminate them. Great strides have been made in understanding how foreign pathogens are identified and responded to, leading to the development of powerful immunological tools, such as vaccines and a myriad of models used to study infectious diseases and processes. However, it is occasionally possible for host tissues themselves to be inappropriately identified as foreign, prompting an immune response that attempts to eliminate the host tissue. The immune system has processes in place, referred to as selection, designed to prevent the development of cells capable of recognizing the self as foreign. -

PI3K Pathway Regulates ER-Dependent Transcription in Breast Cancer Through the Epigenetic Regulator KMT2D
HHS Public Access Author manuscript Author ManuscriptAuthor Manuscript Author Science Manuscript Author . Author manuscript; Manuscript Author available in PMC 2018 March 24. Published in final edited form as: Science. 2017 March 24; 355(6331): 1324–1330. doi:10.1126/science.aah6893. PI3K pathway regulates ER-dependent transcription in breast cancer through the epigenetic regulator KMT2D Eneda Toska1, Hatice U. Osmanbeyoglu2,*, Pau Castel1,3,*, Carmen Chan1, Ronald C. Hendrickson4, Moshe Elkabets1,5, Maura N. Dickler6, Maurizio Scaltriti1,7, Christina S. Leslie2, Scott A. Armstrong8,9, and José Baselga1,6 1Human Oncology and Pathogenesis Program, Memorial Sloan Kettering Cancer Center, 1275 York Avenue, Box 20, New York, NY 10065, USA 2Computational Biology Program, Memorial Sloan Kettering Cancer Center, 1275 York Avenue, Box 460, New York, NY 10065, USA 3Helen Diller Family Comprehensive Cancer Center, University of California–San Francisco, 1450 3rd Street, San Francisco, CA 94158, USA 4Microchemistry and Proteomics Core Laboratory, Memorial Sloan Kettering Cancer Center, New York, NY 10065, USA 5The Shraga Segal Department of Microbiology, Immunology and Genetics, Faculty of Health Sciences, Ben-Gurion University of the Negev, Beer-Sheva 84105, Israel 6Department of Medicine, Memorial Sloan Kettering Cancer Center, New York, NY 10065, USA 7Department of Pathology, Memorial Sloan Kettering Cancer Center, New York, NY 10065, USA 8Cancer Biology and Genetics Program, Memorial Sloan Kettering Cancer Center, New York, NY 10065, USA 9Department of Pediatric Oncology, Dana-Farber Cancer Institute, 450 Brookline Avenue, Boston, MA 02215, USA Abstract Activating mutations in PIK3CA, the gene encoding phosphoinositide-(3)-kinase α (PI3Kα), are frequently found in estrogen receptor (ER)–positive breast cancer. -

UNIVERSITY of CALIFORNIA, IRVINE Combinatorial Regulation By
UNIVERSITY OF CALIFORNIA, IRVINE Combinatorial regulation by maternal transcription factors during activation of the endoderm gene regulatory network DISSERTATION submitted in partial satisfaction of the requirements for the degree of DOCTOR OF PHILOSOPHY in Biological Sciences by Kitt D. Paraiso Dissertation Committee: Professor Ken W.Y. Cho, Chair Associate Professor Olivier Cinquin Professor Thomas Schilling 2018 Chapter 4 © 2017 Elsevier Ltd. © 2018 Kitt D. Paraiso DEDICATION To the incredibly intelligent and talented people, who in one way or another, helped complete this thesis. ii TABLE OF CONTENTS Page LIST OF FIGURES vii LIST OF TABLES ix LIST OF ABBREVIATIONS X ACKNOWLEDGEMENTS xi CURRICULUM VITAE xii ABSTRACT OF THE DISSERTATION xiv CHAPTER 1: Maternal transcription factors during early endoderm formation in 1 Xenopus Transcription factors co-regulate in a cell type-specific manner 2 Otx1 is expressed in a variety of cell lineages 4 Maternal otx1 in the endodermal conteXt 5 Establishment of enhancers by maternal transcription factors 9 Uncovering the endodermal gene regulatory network 12 Zygotic genome activation and temporal control of gene eXpression 14 The role of maternal transcription factors in early development 18 References 19 CHAPTER 2: Assembly of maternal transcription factors initiates the emergence 26 of tissue-specific zygotic cis-regulatory regions Introduction 28 Identification of maternal vegetally-localized transcription factors 31 Vegt and OtX1 combinatorially regulate the endodermal 33 transcriptome iii -

A Flexible Microfluidic System for Single-Cell Transcriptome Profiling
www.nature.com/scientificreports OPEN A fexible microfuidic system for single‑cell transcriptome profling elucidates phased transcriptional regulators of cell cycle Karen Davey1,7, Daniel Wong2,7, Filip Konopacki2, Eugene Kwa1, Tony Ly3, Heike Fiegler2 & Christopher R. Sibley 1,4,5,6* Single cell transcriptome profling has emerged as a breakthrough technology for the high‑resolution understanding of complex cellular systems. Here we report a fexible, cost‑efective and user‑ friendly droplet‑based microfuidics system, called the Nadia Instrument, that can allow 3′ mRNA capture of ~ 50,000 single cells or individual nuclei in a single run. The precise pressure‑based system demonstrates highly reproducible droplet size, low doublet rates and high mRNA capture efciencies that compare favorably in the feld. Moreover, when combined with the Nadia Innovate, the system can be transformed into an adaptable setup that enables use of diferent bufers and barcoded bead confgurations to facilitate diverse applications. Finally, by 3′ mRNA profling asynchronous human and mouse cells at diferent phases of the cell cycle, we demonstrate the system’s ability to readily distinguish distinct cell populations and infer underlying transcriptional regulatory networks. Notably this provided supportive evidence for multiple transcription factors that had little or no known link to the cell cycle (e.g. DRAP1, ZKSCAN1 and CEBPZ). In summary, the Nadia platform represents a promising and fexible technology for future transcriptomic studies, and other related applications, at cell resolution. Single cell transcriptome profling has recently emerged as a breakthrough technology for understanding how cellular heterogeneity contributes to complex biological systems. Indeed, cultured cells, microorganisms, biopsies, blood and other tissues can be rapidly profled for quantifcation of gene expression at cell resolution. -

Context-Dependent Mir-21 Regulation of TLR7-Mediated Autoimmune and Foreign Antigen
bioRxiv preprint doi: https://doi.org/10.1101/2021.03.12.435182; this version posted March 13, 2021. The copyright holder for this preprint (which was not certified by peer review) is the author/funder. All rights reserved. No reuse allowed without permission. 1 Context-dependent miR-21 regulation of TLR7-mediated autoimmune and foreign antigen 2 driven antibody-forming cell and germinal center responses 3 Stephanie L. Schell1,2, Kristen N. Bricker1,2, Adam J. Fike1, Sathi Babu Chodisetti1, Phillip P. 4 Domeier3, Nicholas M. Choi1, Melinda J. Fasnacht1, Sara A. Luckenbill1, Steven F. Ziegler3, and 5 Ziaur S.M. Rahman1* 6 1Department of Microbiology and Immunology, Pennsylvania State University College of 7 Medicine, Hershey, PA 17033 8 2S.L.S and K.N.B contributed equally to this work 9 3Immunology Program, Benaroya Research Institute, Seattle, WA 98101 10 *Address correspondence and reprint requests to Dr. Ziaur Rahman, Department of 11 Microbiology and Immunology, H107, Pennsylvania State University College of Medicine, 500 12 University Drive, Hershey, PA 17033. 13 Email: [email protected] 14 Ph: (717) 531-0003 x287896; Fax: (717) 531-6522 15 Running title: miR-21 regulation of AFC, GC and Ab responses 16 17 Abbreviations: Ab, antibody; AFC, antibody forming cell; ANA, anti-nuclear antibody; BCR, 18 B cell receptor; GC, germinal center; IMQ, imiquimod; GO, gene ontology; GSEA, gene set 19 enrichment analysis; miR, microRNA; SLE, systemic lupus erythematosus; SmRNP, Smith 20 antigen/ribonucleoprotein; Tfh, T follicular helper cell; Tfr, T follicular regulatory cell; TI-IFN, 21 Type I interferon 22 23 24 bioRxiv preprint doi: https://doi.org/10.1101/2021.03.12.435182; this version posted March 13, 2021. -

Curcumin Alters Gene Expression-Associated DNA Damage, Cell Cycle, Cell Survival and Cell Migration and Invasion in NCI-H460 Human Lung Cancer Cells in Vitro
ONCOLOGY REPORTS 34: 1853-1874, 2015 Curcumin alters gene expression-associated DNA damage, cell cycle, cell survival and cell migration and invasion in NCI-H460 human lung cancer cells in vitro I-TSANG CHIANG1,2, WEI-SHU WANG3, HSIN-CHUNG LIU4, SU-TSO YANG5, NOU-YING TANG6 and JING-GUNG CHUNG4,7 1Department of Radiation Oncology, National Yang‑Ming University Hospital, Yilan 260; 2Department of Radiological Technology, Central Taiwan University of Science and Technology, Taichung 40601; 3Department of Internal Medicine, National Yang‑Ming University Hospital, Yilan 260; 4Department of Biological Science and Technology, China Medical University, Taichung 404; 5Department of Radiology, China Medical University Hospital, Taichung 404; 6Graduate Institute of Chinese Medicine, China Medical University, Taichung 404; 7Department of Biotechnology, Asia University, Taichung 404, Taiwan, R.O.C. Received March 31, 2015; Accepted June 26, 2015 DOI: 10.3892/or.2015.4159 Abstract. Lung cancer is the most common cause of cancer CARD6, ID1 and ID2 genes, associated with cell survival and mortality and new cases are on the increase worldwide. the BRMS1L, associated with cell migration and invasion. However, the treatment of lung cancer remains unsatisfactory. Additionally, 59 downregulated genes exhibited a >4-fold Curcumin has been shown to induce cell death in many human change, including the DDIT3 gene, associated with DNA cancer cells, including human lung cancer cells. However, the damage; while 97 genes had a >3- to 4-fold change including the effects of curcumin on genetic mechanisms associated with DDIT4 gene, associated with DNA damage; the CCPG1 gene, these actions remain unclear. Curcumin (2 µM) was added associated with cell cycle and 321 genes with a >2- to 3-fold to NCI-H460 human lung cancer cells and the cells were including the GADD45A and CGREF1 genes, associated with incubated for 24 h. -

PBX1 Acts As Terminal Selector for Olfactory Bulb Dopaminergic Neurons
© 2020. Published by The Company of Biologists Ltd | Development (2020) 147, dev186841. doi:10.1242/dev.186841 RESEARCH ARTICLE PBX1 acts as terminal selector for olfactory bulb dopaminergic neurons Laura Remesal1,*,‡, Isabel Roger-Baynat1,‡, Laura Chirivella1, Miren Maicas1, Rebeca Brocal-Ruiz1, Ana Pérez-Villalba2, Carme Cucarella3, Marta Casado3 and Nuria Flames1,§ ABSTRACT expressed in the nervous system throughout the various stages of Neuronal specification is a protracted process that begins with the differentiation, from progenitors to mature neurons (Delile et al., commitment of progenitor cells and culminates with the generation of 2019; Mayer et al., 2018; Mi et al., 2018; Nowakowski et al., 2017). mature neurons. Many transcription factors are continuously In only a few cases have the specific roles of these TFs in progenitors expressed during this process but it is presently unclear how these and postmitotic neurons been determined, such as the diverse factors modify their targets as cells transition through different stages functions of Nkx2.1 in the subpallium (Magno et al., 2011; Nóbrega- of specification. In olfactory bulb adult neurogenesis, the transcription Pereira et al., 2008; Sussel et al., 1999), or the specific roles of Fezf2, factor PBX1 controls neurogenesis in progenitor cells and the survival Brn1/2 and Cux1/2 at particular differentiation stages in the pallium of migrating neuroblasts. Here, we show that, at later differentiation (Cubelos et al., 2008; Guo et al., 2013; Iulianella et al., 2008; Lodato stages, PBX1 also acts as a terminal selector for the dopaminergic et al., 2014; McEvilly et al., 2002; Sugitani et al., 2002). However, neuron fate. -

Targeted Exome Sequencing Identifies PBX1 As Involved
BASIC RESEARCH www.jasn.org Targeted Exome Sequencing Identifies PBX1 as Involved in Monogenic Congenital Anomalies of the Kidney and Urinary Tract † ‡ | | Laurence Heidet,* Vincent Morinière,* Charline Henry,§ Lara De Tomasi,§ ¶ | | ‡†† Madeline Louise Reilly,§ ¶ Camille Humbert,§ Olivier Alibeu,** Cécile Fourrage, †† †† †† Christine Bole-Feysot,** Patrick Nitschké, Frédéric Tores, Marc Bras, |‡‡ || Marc Jeanpierre, Christine Pietrement,§§ Dominique Gaillard, Marie Gonzales,¶¶ ††† ‡‡‡ ||| Robert Novo,*** Elise Schaefer, Joëlle Roume, Jelena Martinovic,§§§ Valérie Malan, † | | ‡ | | Rémi Salomon,* § Sophie Saunier,§ Corinne Antignac, § and Cécile Jeanpierre§ *Assistance Publique - Hôpitaux de Paris, Centre de référence des Maladies Rénales Héréditaires de l’Enfant et de l’Adulte, Paris, France; †Assistance Publique - Hôpitaux de Paris, Service de Néphrologie Pédiatrique, ‡Assistance Publique - Hôpitaux de Paris, Département de Génétique, and |||Assistance Publique - Hôpitaux de Paris, Service de Cytogénétique, Hôpital Universitaire Necker- Enfants malades, Paris, France; §Institut National de la Santé et de la Recherche Médicale Unité Mixte de Recherche 1163, Laboratory of Hereditary Kidney Diseases, **Genomic Platform, Institut National de la Santé et de la Recherche Médicale Unité Mixte de Recherche 1163, Paris Descartes Sorbonne Paris Cité University, and ††Bioinformatic Plateform, Paris Descartes Sorbonne Paris Cité University, Imagine Institute, Paris, France; |Paris Descartes Sorbonne Paris Cité University, Paris, France; ¶Paris -
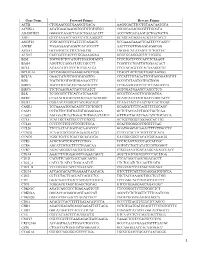
Gene Name Forward Primer Reverse Primer ACTB
Gene Name Forward Primer Reverse Primer ACTB CTGGAACGGTGAAGGTGACA AAGGGACTTCCTGTAACAATGCA ACVRL1 ACATGAAGAAGGTGGTGTGTGTGG CGGGCAGAGGGGTTTGGGTA ADAMDEC1 GGGGCCAGACTACACTGAAACATT ACCCGTCACAAGTACTGATGCTG AHI1 GTCCAAAACTACCCCATCAAGGCT GCAGCACAGGAACGTATCACCT ANGPT2 TGGCAGCGTTGATTTTCAGAGG GCGAAACAAACTCATTTCCCAGCC ANPEP TGAAGAAGCAGGTCACACCCCT AACTCCGTTGGAGCAGGCGG APOA1 GCCGTGCTCTTCCTGACGG TGGGACACATAGTCTCTGCCGC ATXN7 CACCGCCCACTCTGGAAAAGAA GGGTGCAGGGCTTCTTGGTG B2M TGCTGTCTCCATGTTTGATGTATCT TCTCTGCTCCCCACCTCTAAGT BAG4 AGGTTCCAGGATATCCGCCTT TCGGTCCTGATTGTGGAACACT BCL2 ACAACATCGCCCTGTGGATGA CCGTACAGTTCCACAAAGGCAT BCL2L14 GCTCAGGGTCAAAGGACGTTGG TCAGCTACTCGGTTGGCAATGG BCL7A GAACCATGTCGGGCAGGTCG CCCATTTGTAGATTCGTAGGGATGTGT BIN1 TGCTGTCGTGGTGGAGACCTTC GCCGTGTAGTCGTGCTGGG BIRC3 TGCTATCCACATCAGACAGCCC TCTGAATGGTCTTCTCCAGGTTCA BIRC5 TTCTCAAGGACCACCGCATCT AGTGGATGAAGCCAGCCTCG BLK TCGGGGTCTTCACCATCAAAGC GCGCTCCAGGTTGCGGATGA BTRC CCAAATGTGTCATTACCAACATGGGC GCAGCACATAGTGATTTGGCATCC BUB3 CGGAACATGGGTTACGTGCAGC CCAAATACTCAACTGCCACTCGGC CAGE1 TCCAAAATGCACAGTCTTCTGGCT GGAGGCTCTTCAGTTTTTGCAGC CASP1 CCTGTTCCTGTGATGTGGAGGAAA GCTCTACCATCTGGCTGCTCAA CASP3 AGCGAATCAATGGACTCTGGAATATCC GTTTGCTGCATCGACATCTGTACCA CCL5 TCATTGCTACTGCCCTCTGCG ACTGCTGGGTTGGAGCACTTG CCL18 CCCTCCTTGTCCTCGTCTGCA GCACTGGGGGCTGGTTTCAG CCL26 TTCCAATACAGCCACAAGCCCC GGATGGGTACAGACTTTCTTGCCTC CCND2 TCAAGTGCGTGCAGAAGGACAT CTTCGCACTTCTGTTCCTCACA CCND3 TGGCTGCTGTGATTGCACATGA GATGGCGGGTACATGGCAAAGG CCR3 ACGCTGCTCTGCTTCCTGG TCCTCAGTTCCCCACCATCGC CCR4 AGCATCGTGCTTCCTGAGCAA GGTGTCTGCTATATCCGTGGGGT CCR7 AGACAGGGGTAGTGCGAGGC