Device Architecture and Materials for Organic Light-Emitting Devices Sarah Schols
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

17 Electronics Assembly Basic Expe- Riments with Breadboard
118.381 17 Electronics Assembly Basic Expe- riments with BreadboardTools Required: Stripper Side Cutters Please Note! The Opitec Range of projects is not intended as play toys for young children. They are teaching aids for young people learning the skills of craft, design and technology. These projects should only be undertaken and operated with the guidance of a fully qualified adult. The finished pro- jects are not suitable to give to children under 3 years old. Some parts can be swallowed. Danger of suffocation! Article List Quantity Size (mm) Designation Part-No. Plug-in board/ breadboard 1 83x55 Plug-in board 1 Loudspeaker 1 Loudspeaker 2 Blade receptacle 2 Connection battery 3 Resistor 120 Ohm 2 Resistor 4 Resistor 470 Ohm 1 Resistor 5 Resistor 1 kOhm 1 Resistor 6 Resistor 2,7 kOhm 1 Resistor 7 Resistor 4,7 kOhm 1 Resistor 8 Resistor 22 kOhm 1 Resistor 9 Resistor 39 kOhm 1 Resistor 10 Resistor 56 kOhm 1 Resistor 11 Resistor 1 MOhm 1 Resistor 12 Photoconductive cell 1 Photoconductive cell 13 Transistor BC 517 2 Transistor 14 Transistor BC 548 2 Transistor 15 Transistor BC 557 1 Transistor 16 Capacitor 4,7 µF 1 Capacitor 17 Elko 22µF 2 Elko 18 elko 470µF 1 Elko 19 LED red 1 LED 20 LED green 1 LED 21 Jumper wire, red 1 2000 Jumper Wire 22 1 Instruction 118.381 17 Electronics Assembly Basic Experiments with Breadboard General: How does a breadboard work? The breadboard also called plug-in board - makes experimenting with electronic parts immensely easier. The components can simply be plugged into the breadboard without soldering them. -

10-12/Electronic Components April 8, 2020 10-12/Digital Electronics Lesson: 4/8/2020
10-12 PLTW Engineering 10-12/Electronic Components April 8, 2020 10-12/Digital Electronics Lesson: 4/8/2020 Objective/Learning Target: Students will be able to read the resistance value in Ohms of a common resistor and identify common electronics components. Resistors •. Resistors are an electronic component that resist the flow of current in an electrical circuit • They are measured in Ohms (Ω) • The different colored bands represent how much current flow that specific resistor can oppose • They are useful for reducing current before indicators like LED lights and buzzers. Resistors To read the resistors we use a Color Code Table 1. Starting at the end with the band closest to the end, we match the color with the number on the chart for the first 2 bands. 2. The 3rd band is designated as the multiplier. This indicates how many zeros to add to the number you got reading the first to bands. 3. The 4th band is designated as the tolerance. This tells us how much the actual resistance value may vary from what is represented on the chart. Resistors Lets do an example using the Color Code Table Starting at the end with the band closest to the end, we see the 1st band is Red, 2nd band is Violet. So, we have 27 so far. Next is the multiplier. In this case Brown, or 1. So we only add 1 zero. This puts the value of the resistor at 270 ohms. Finally, the tolerance is Gold or +-5%. So overall, the value of this resistor is 270Ω +-5% Capacitors • .Another common electronic component are capacitors. -

Basic Electronic Components
BASIC ELECTRONIC COMPONENTS MODEL ECK-10 Resistors Capacitors Coils Others Transformers Semiconductors Instruction Manual by Arthur F. Seymour MSEE It is the intention of this course to teach the fundamental operation of basic electronic components by comparison to drawings of equivalent mechanical parts. It must be understood that the mechanical circuits would operate much slower than their electronic counterparts and one-to-one correlation can never be achieved. The comparisons will, however, give an insight to each of the fundamental electronic components used in every electronic product. ElencoTM Electronics, Inc. Copyright © 2004, 1994 ElencoTM Electronics, Inc. Revised 2004 REV-G 753254 RESISTORS RESISTORS, What do they do? The electronic component known as the resistor is Electrons flow through materials when a pressure best described as electrical friction. Pretend, for a (called voltage in electronics) is placed on one end moment, that electricity travels through hollow pipes of the material forcing the electrons to “react” with like water. Assume two pipes are filled with water each other until the ones on the other end of the and one pipe has very rough walls. It would be easy material move out. Some materials hold on to their to say that it is more difficult to push the water electrons more than others making it more difficult through the rough-walled pipe than through a pipe for the electrons to move. These materials have a with smooth walls. The pipe with rough walls could higher resistance to the flow of electricity (called be described as having more resistance to current in electronics) than the ones that allow movement than the smooth one. -

Basic Electronic Components
ECK-10_REV-O_091416.qxp_ECK-10 9/14/16 2:49 PM Page 1 BASIC ELECTRONIC COMPONENTS MODEL ECK-10 Coils Capacitors Resistors Others Transformers (not included) Semiconductors Instruction Manual by Arthur F. Seymour MSEE It is the intention of this course to teach the fundamental operation of basic electronic components by comparison to drawings of equivalent mechanical parts. It must be understood that the mechanical circuits would operate much slower than their electronic counterparts and one-to-one correlation can never be achieved. The comparisons will, however, give an insight to each of the fundamental electronic components used in every electronic product. ® ELENCO ® Copyright © 2016, 1994 by ELENCO Electronics, Inc. All rights reserved. Revised 2016 REV-O 753254 No part of this book shall be reproduced by any means; electronic, photocopying, or otherwise without written permission from the publisher. ECK-10_REV-O_091416.qxp_ECK-10 9/14/16 2:49 PM Page 2 RESISTORS RESISTORS, What do they do? The electronic component known as the resistor is Electrons flow through materials when a pressure best described as electrical friction. Pretend, for a (called voltage in electronics) is placed on one end moment, that electricity travels through hollow pipes of the material forcing the electrons to “react” with like water. Assume two pipes are filled with water each other until the ones on the other end of the and one pipe has very rough walls. It would be easy material move out. Some materials hold on to their to say that it is more difficult to push the water electrons more than others making it more difficult through the rough-walled pipe than through a pipe for the electrons to move. -

Iiic Store.Category.Electronic Component.Subassembly Part.Power Supplies.Switching Power Supply
800WParallel(N+1)WithPFCFunction SCP-800 series Features : AC input 180~260VAC only PF> 0.98@ 230VAC Protections: Short circuit / Overload / Over voltage / Over temperature Built in remote sense function Built-in remote ON-OFF control Built-in power good signal output Built-in parallel operation function(N+1) Can adjust from 20~100% output voltage by external control 1-5V Forced air cooling by built-in DC fan 3 years warranty SPECIFICATION ORDERNO. SCP-800-09 SCP-800-12 SCP-800-15 SCP-800-18 SCP-800-24 SCP-800-36 SCP-800-48 SCP-800-60 SAFETY MODEL NO. 800S-P009 800S-P012 800S-P015 800S-P018 800S-P024 800S-P036 800S-P048 800S-P060 DCVOLTAGE 9V 12V 15V 18V 24V 36V 48V 60V RATEDCURRENT 88A 66A 53A 44.4A 33A 22.2A 16A 13A CURRENTRANGE 0~88A 0~66A 0~53A 0~44.4A 0~33A 0~22.2A 0~16A 0~13A RATEDPOWER 792W 792W 795W 799W 792W 799W 768W 780W OUTPUT RIPPLE&NOISE(max.) Note.2 90mVp-p 120mVp-p 150mVp-p 180mVp-p 240mVp-p 360mVp-p 480mVp-p 500mVp-p VOLTAGE ADJ.RANGE 3.0% Typicaladjustmentbypotentiometer20%~100%adjustmentby1~5VDCexternalcontrol VOLTAGETOLERANCE Note.3 1.5% 1.0% 1.0% 1.0% 1.0% 1.0% 1.0% 1.0% LINEREGULATION 0.5% 0.5% 0.5% 0.5% 0.5% 0.5% 0.5% 0.5% LOADREGULATION 1.0% 0.5% 0.5% 0.5% 0.5% 0.5% 0.5% 0.5% SETUP,RISE,HOLDUP TIME 800ms,400ms,12msatfullload VOLTAGERANGE 180~260VAC260~370VDCseethederatingcurve FREQUENCY RANGE 47~63Hz POWERFACTOR >0.98/230VAC INPUT EFFICIENCY (Typ.) 83% 84% 85% 86% 88% 88% 89% 90% ACCURRENT 5.0A /230VAC INRUSHCURRENT(max.) 60A /230VAC LEAKAGECURRENT(max.) 3.5mA /240VAC 105~115%ratedoutputpower OVERLOAD Note.4 Protectiontype: Currentlimiting,delayshutdowno/pvoltage,re-powerontorecover 110~135% Followtooutputsetuppoint PROTECTION OVERVOLTAGE Protectiontype:Shutdowno/pvoltage,re-powerontorecover >100 /measurebyheatsink,neartransformer OVERTEMPERATURE Protectiontype:Shutdowno/pvoltage, recoversautomaticallyaftertemperaturegoesdown WORKINGTEMP. -

The Designer's Guide to Instrumentation Amplifiers
A Designer’s Guide to Instrumentation Amplifiers 3 RD Edition www.analog.com/inamps A DESIGNER’S GUIDE TO INSTRUMENTATION AMPLIFIERS 3RD Edition by Charles Kitchin and Lew Counts i All rights reserved. This publication, or parts thereof, may not be reproduced in any form without permission of the copyright owner. Information furnished by Analog Devices, Inc. is believed to be accurate and reliable. However, no responsibility is assumed by Analog Devices, Inc. for its use. Analog Devices, Inc. makes no representation that the interconnec- tion of its circuits as described herein will not infringe on existing or future patent rights, nor do the descriptions contained herein imply the granting of licenses to make, use, or sell equipment constructed in accordance therewith. Specifications and prices are subject to change without notice. ©2006 Analog Devices, Inc. Printed in the U.S.A. G02678-15-9/06(B) ii TABLE OF CONTENTS CHAPTER I—IN-AMP BASICS ...........................................................................................................1-1 INTRODUCTION ...................................................................................................................................1-1 IN-AMPS vs. OP AMPS: WHAT ARE THE DIFFERENCES? ..................................................................1-1 Signal Amplification and Common-Mode Rejection ...............................................................................1-1 Common-Mode Rejection: Op Amp vs. In-Amp .....................................................................................1-3 -

Controlling Nanostructure in Inkjet Printed Organic Transistors for Pressure Sensing Applications
nanomaterials Article Controlling Nanostructure in Inkjet Printed Organic Transistors for Pressure Sensing Applications Matthew J. Griffith 1,2,* , Nathan A. Cooling 1, Daniel C. Elkington 1, Michael Wasson 1 , Xiaojing Zhou 1, Warwick J. Belcher 1 and Paul C. Dastoor 1 1 Centre for Organic Electronics, University of Newcastle, University Drive, Callaghan, NSW 2308, Australia; [email protected] (N.A.C.); [email protected] (D.C.E.); [email protected] (M.W.); [email protected] (X.Z.); [email protected] (W.J.B.); [email protected] (P.C.D.) 2 School of Aeronautical, Mechanical and Mechatronic Engineering, University of Sydney, Camperdown, NSW 2006, Australia * Correspondence: matthew.griffi[email protected] Abstract: This work reports the development of a highly sensitive pressure detector prepared by inkjet printing of electroactive organic semiconducting materials. The pressure sensing is achieved by incorporating a quantum tunnelling composite material composed of graphite nanoparticles in a rubber matrix into the multilayer nanostructure of a printed organic thin film transistor. This printed device was able to convert shock wave inputs rapidly and reproducibly into an inherently amplified electronic output signal. Variation of the organic ink material, solvents, and printing speeds were shown to modulate the multilayer nanostructure of the organic semiconducting and dielectric layers, enabling tuneable optimisation of the transistor response. The optimised printed device exhibits Citation: Griffith, M.J.; Cooling, rapid switching from a non-conductive to a conductive state upon application of low pressures whilst N.A.; Elkington, D.C.; Wasson, M.; operating at very low source-drain voltages (0–5 V), a feature that is often required in applications Zhou, X.; Belcher, W.J.; Dastoor, P.C. -

Light-Emitting Diode - Wikipedia, the Free Encyclopedia
Light-emitting diode - Wikipedia, the free encyclopedia http://en.wikipedia.org/wiki/Light-emitting_diode From Wikipedia, the free encyclopedia A light-emitting diode (LED) (pronounced /ˌɛl iː ˈdiː/[1]) is a semiconductor Light-emitting diode light source. LEDs are used as indicator lamps in many devices, and are increasingly used for lighting. Introduced as a practical electronic component in 1962,[2] early LEDs emitted low-intensity red light, but modern versions are available across the visible, ultraviolet and infrared wavelengths, with very high brightness. When a light-emitting diode is forward biased (switched on), electrons are able to recombine with holes within the device, releasing energy in the form of photons. This effect is called electroluminescence and the color of the light (corresponding to the energy of the photon) is determined by the energy gap of Red, green and blue LEDs of the 5mm type 2 the semiconductor. An LED is usually small in area (less than 1 mm ), and Type Passive, optoelectronic integrated optical components are used to shape its radiation pattern and assist in reflection.[3] LEDs present many advantages over incandescent light sources Working principle Electroluminescence including lower energy consumption, longer lifetime, improved robustness, Invented Nick Holonyak Jr. (1962) smaller size, faster switching, and greater durability and reliability. LEDs powerful enough for room lighting are relatively expensive and require more Electronic symbol precise current and heat management than compact fluorescent lamp sources of comparable output. Pin configuration Anode and Cathode Light-emitting diodes are used in applications as diverse as replacements for aviation lighting, automotive lighting (particularly indicators) and in traffic signals. -

Physics 197 Lab 12: Planck's Constant from LED Spectra
Physics 197 Lab 12: Planck’s Constant from LED Spectra Equipment: Item Part # Qty # of Total Storage Qty Qty per Teams Qty Location Set Put Team Needed Out Back Red Tide Spectrometer Vernier V-Spec 1 6 6 Computer with Logger Pro 1 6 6 Optical Fiber Assembly For Red Tide 1 6 6 Ring Stand and Clamp 1 6 6 USB Cable – Red Tide to Computer 1 6 6 Planck’s constant LED apparatus Esco 1 6 6 DMM and probes for voltage 1 6 6 DMM and probes for current 1 6 6 Layouts: Figure 1, LED Planck’s Constant Setup Figure 2, Measurement of LED Voltage at Emission Threshold Summary: In this lab, students will determine a value for Planck’s constant by measuring the emission wavelength of 7 LED’s along with the threshold voltage at which they first turn on. The LED’s range in wavelength from 470nm to 940 nm. The emission spectra will be observed with a Red Tide Spectrometer coupled to the LED’s with a fiber optic cable which can be positioned right above the LED. The peak wavelength of the emission curve will be measured using the spectrometer. The voltage to the LED can be varied, and a voltmeter will be used to measure the voltage at which the LED just starts emitting, which should be close to the bandgap. The energy of the emitted photons should be close to the bandgap voltage multiplied by the electron charge, and should also be equal to Planck’s constant multiplied by the photon frequency, allowing for a calculation of Planck’s constant. -

White Paper Basic Switches
White Paper Issued By: Issue Date: March 2019 Basics of Basic Switches General Knowledge of Basic Switches Introduction Sensors in the past were considered one of the common electronic components used as complements to the functions and performance of electric appliances, machines and mechanical equipment. Today, with the advent of IoT, sensors and other electronic devices directly connect to the Internet and will play a vital role in the future IoT society - not limited to merely an electronic component that plays a complementary role. “Basic switch” is a mechanical sensor that detects (senses) the presence or absence of an object as well as its position by activating a plunger when the object physically comes into contact. With the emerging IoT, Omron’s basic switches have been widely adopted for position detection installed in customers’ newly developed electric appliances, machines and mechanical equipment. Furthermore, electrical signals outputted from the basic switch that indicates normal and abnormal conditions when detecting positions can be transmitted to remote monitoring systems over the Internet. Definition The following is the definition of a basic switch provided by the Nippon Electric Control Equipment Industries Association (NECA). “A miniature switch, also known as snap-action switch, achieves a very small contact separation and the contacts are enclosed in a case in which these contacts open and close according to the specified movement and force. An actuator is equipped outside the case ”(Figure 1) With a snap-action mechanism, the contacts will instantaneously switch at a specific operating position regardless of its speed. The mechanism allows the basic switch to operate using a specified movement and force. -

Guidelines for Unit Packing for Integrated Circuits - Tubes (Rails)
Guidelines for Unit Packing for Integrated Circuits - Tubes (Rails) NIGP 102.00 SEPTEMBER 1992 NATIONAL ELECTRONIC DISTRIBUTORS ASSOCIATION 1111 Alderman Drive, Suite 400 Alpharetta, GA 30005-4143 678-393-9990/678-393-9998 fax www.nedassoc.org NOTICE NEDA Industry Guidelines and Publications contain material that has been prepared, progressively reviewed, and approved through various NEDA sponsored industry task forces comprised of NEDA member distributors and manufacturers and subsequently reviewed and approved by the NEDA Board of Directors. NEDA Industry Guidelines and Publications are designed to serve the public interest including electronic component distributors through the promotion of standardized practices between manufacturers and distributors resulting in improved efficiency, profitability and product quality. Existence of such guidelines shall not in any respect preclude any member or nonmember of NEDA from selling or manufacturing products not in conformance to such guidelines, nor shall the existence of such guidelines preclude their voluntary use by those other than NEDA members, whether the guideline is to be used either domestically or internationally. NEDA does not assume any liability or obligation whatever to parties adopting NEDA industry Guidelines and Publications. Inquiries, comments and suggestions relative to the content of this NEDA Industry Guideline should be addressed to NEDA headquarters. Published by NATIONAL ELECTRONIC DISTRIBUTORS ASSOCIATION 1111 Alderman Drive, Suite 400 Alpharetta, GA 30005 678.393.9990/678.393.9998 fax Copyright 1992 Printed in U.S.A. All rights reserved NEDA Guidelines for Unit Packing for Integrated Circuits - Tubes (Rails) Introduction Growing global competition for the industries which comprise the predominant users of electronic components, particularly semiconductor products, is resulting in a continuous evolution in customers' practices and their vendor performance expectations. -
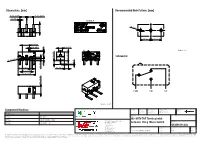
463091691402 Datasheet WS-MITV THT Terminal with Actuator, 150 G Micro Switch
Dimensions: [mm] Recommended Hole Pattern: [mm] 5,08 ±0,15 5,08 ±0,15 1,0 ±0,1 detail A O 1,2 6,2 ±0,35 5,08 ±0,1 5,08 ±0,1 5,2 ±0,2 1,2 ±0,3 2,9 ±0,3 PT A Scale - 3:1 OP Schematic: 0,3 2,8 ±0,2 O 2,0 ± 6,5 ±0,15 0,4 1,5 O 2,2 ±0,3 2,0 ±0,3 3,1 ±0,2 6,5 ±0,1 12,8 ±0,15 5,8 ±0,15 COM NO NC Scale - 2,5:1 CHECKED REVISION DATE (YYYY-MM-DD) GENERAL TOLERANCE PROJECTION Component Marking: METHOD ICh 002.000 2021-04-19 DIN ISO 2768-1m Marking 1: DESCRIPTION 1st line WE-MITV 2nd line 3A GP 125VAC ENEC WS-MITV THT Terminal with Würth Elektronik eiSos GmbH & Co. KG ORDER CODE rd EMC & Inductive Solutions Actuator, 150 g Micro Switch 3 line 2A 30VDC UL Max-Eyth-Str. 1 74638 Waldenburg 463091691402 Germany Tel. +49 (0) 79 42 945 - 0 SIZE/TYPE BUSINESS UNIT STATUS PAGE www.we-online.com [email protected] 12.8 x 5.8 mm Right Angle Operation eiCan Valid 1/9 This electronic component has been designed and developed for usage in general electronic equipment only. This product is not authorized for use in equipment where a higher safety standard and reliability standard is especially required or where a failure of the product is reasonably expected to cause severe personal injury or death, unless the parties have executed an agreement specifically governing such use.