Download Thesis
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

(19) United States (12) Patent Application Publication (10) Pub
US 20130289061A1 (19) United States (12) Patent Application Publication (10) Pub. No.: US 2013/0289061 A1 Bhide et al. (43) Pub. Date: Oct. 31, 2013 (54) METHODS AND COMPOSITIONS TO Publication Classi?cation PREVENT ADDICTION (51) Int. Cl. (71) Applicant: The General Hospital Corporation, A61K 31/485 (2006-01) Boston’ MA (Us) A61K 31/4458 (2006.01) (52) U.S. Cl. (72) Inventors: Pradeep G. Bhide; Peabody, MA (US); CPC """"" " A61K31/485 (201301); ‘4161223011? Jmm‘“ Zhu’ Ansm’ MA. (Us); USPC ......... .. 514/282; 514/317; 514/654; 514/618; Thomas J. Spencer; Carhsle; MA (US); 514/279 Joseph Biederman; Brookline; MA (Us) (57) ABSTRACT Disclosed herein is a method of reducing or preventing the development of aversion to a CNS stimulant in a subject (21) App1_ NO_; 13/924,815 comprising; administering a therapeutic amount of the neu rological stimulant and administering an antagonist of the kappa opioid receptor; to thereby reduce or prevent the devel - . opment of aversion to the CNS stimulant in the subject. Also (22) Flled' Jun‘ 24’ 2013 disclosed is a method of reducing or preventing the develop ment of addiction to a CNS stimulant in a subj ect; comprising; _ _ administering the CNS stimulant and administering a mu Related U‘s‘ Apphcatlon Data opioid receptor antagonist to thereby reduce or prevent the (63) Continuation of application NO 13/389,959, ?led on development of addiction to the CNS stimulant in the subject. Apt 27’ 2012’ ?led as application NO_ PCT/US2010/ Also disclosed are pharmaceutical compositions comprising 045486 on Aug' 13 2010' a central nervous system stimulant and an opioid receptor ’ antagonist. -

Enhanced Reporting
Drug Profile, Targeted with Interpretation by Tandem Mass Spectrometry and Enzyme Immunoassay, Urine Patient: PAIN HYB 2, 2009288 | Date of Birth: | Gender: M | Physician: DR T. TEST Patient Identifiers: | Visit Number (FIN): Drug Analyte Result Cutoff Notes Meperidine metabolite Not Detected 50 ng/mL normeperidine Tapentadol Not Detected 100 ng/mL --Tapentadol-o-sulfate Not Detected 200 ng/mL tapentadol metabolite AMPHETAMINE-LIKE, MASS SPEC Amphetamine Present 50 ng/mL eg, Vyvanse; also a metabolite of methamphetamine Methamphetamine Present 200 ng/mL d- and l- isomers are not distinguished by this test; may reflect Vicks inhaler, Desoxyn, Selegiline, or illicit source MDMA - Ecstasy Not Detected 200 ng/mL MDA Not Detected 200 ng/mL also a metabolite of MDMA and MDEA MDEA - Eve Not Detected 200 ng/mL Phentermine Not Detected 100 ng/mL Methylphenidate Not Detected 100 ng/mL eg, Ritalin, Dexmethylphenidate, Focalin, Concerta BENZODIAZEPINE-LIKE, MASS SPEC Alprazolam Not Detected 40 ng/mL eg, Xanax --Alpha-hydroxyalprazolam Not Detected 20 ng/mL alprazolam metabolite Clonazepam Not Detected 20 ng/mL eg, Klonopin --7-aminoclonazepam Not Detected 40 ng/mL clonazepam metabolite Diazepam Not Detected 50 ng/mL eg, Valium Nordiazepam Not Detected 50 ng/mL metabolite of chlordiazepoxide (Librium), clorazepate (Tranxene), diazepam, halazepam (Alapryl), prazepam (Centrax) and others Oxazepam Not Detected 50 ng/mL eg, Serax; also metabolite of nordiazepam and temazepam Temazepam Not Detected 50 ng/mL eg, Restoril; also a metabolite of diazepam Lorazepam Not Detected 60 ng/mL eg, Ativan Midazolam Not Detected 20 ng/mL eg, Versed Zolpidem Present 20 ng/mL eg, Ambien Reference interval Creatinine value (mg/dL) 200.0 20.0 - 400.0 mg/dL Patient: PAIN HYB 2, 2009288 ARUP Accession: 21-048-107698 4848 Chart continues on following page(s) ARUP Enhanced Reporting | February 17, 2021 | page 4 of 5 Drug Profile, Targeted with Interpretation by Tandem Mass Spectrometry and Enzyme Immunoassay, Urine Patient: PAIN HYB 2, 2009288 | Date of Birth | Gender: M | Physician: DR T. -

Amphetamine/Dextroamphetamine IR Generic
GEORGIA MEDICAID FEE-FOR-SERVICE STIMULANT AND RELATED AGENTS PA SUMMARY Preferred Non-Preferred Amphetamine/dextroamphetamine IR generic Adzenys ER (amphetamine ER oral suspension) Armodafinil generic Adzenys XR (amphetamine ER dispersible tab) Atomoxetine generic Amphetamine/dextroamphetamine ER (generic Concerta (methylphenidate ER/SA) Adderall XR) Dextroamphetamine IR tablets generic Aptensio XR (methylphenidate ER) Focalin (dexmethylphenidate) Clonidine ER generic Focalin XR (dexmethylphenidate ER) Cotempla XR (methylphenidate ER disintegrating Guanfacine ER generic tablet) Methylin oral solution (methylphenidate) Daytrana (methylphenidate TD patch) Methylphenidate CD/CR/ER generic by Lannett Desoxyn (methamphetamine) [NDCs 00527-####-##] and Kremers Urban [NDCs Dexmethylphenidate IR generic 62175-####-##] (generic Metadate CD) Dexmethylphenidate ER generic Methylphenidate IR generic Dextroamphetamine ER capsules generic Modafinil generic Dextroamphetamine oral solution generic Quillichew ER (methylphenidate ER chew tabs) Dyanavel XR (amphetamine ER oral suspension) Quillivant XR (methylphenidate ER oral suspension) Evekeo (amphetamine tablets) Vyvanse (lisdexamfetamine) Methamphetamine generic Zenzedi 5 mg, 10 mg IR tablets (dextroamphetamine) Methylphenidate IR chewable tablets generic Methylphenidate ER/SA (generic Concerta) Methylphenidate ER/LA/SR (generic Ritalin LA, Ritalin SR, Metadate ER) Methylphenidate ER/SA 72 mg generic Methylphenidate oral solution generic Mydayis (amphetamine/dextroamphetamine ER) Ritalin LA 10 mg -

Current Topics in Behavioral Neurosciences
Current Topics in Behavioral Neurosciences Series Editors Mark A. Geyer, La Jolla, CA, USA Bart A. Ellenbroek, Wellington, New Zealand Charles A. Marsden, Nottingham, UK For further volumes: http://www.springer.com/series/7854 About this Series Current Topics in Behavioral Neurosciences provides critical and comprehensive discussions of the most significant areas of behavioral neuroscience research, written by leading international authorities. Each volume offers an informative and contemporary account of its subject, making it an unrivalled reference source. Titles in this series are available in both print and electronic formats. With the development of new methodologies for brain imaging, genetic and genomic analyses, molecular engineering of mutant animals, novel routes for drug delivery, and sophisticated cross-species behavioral assessments, it is now possible to study behavior relevant to psychiatric and neurological diseases and disorders on the physiological level. The Behavioral Neurosciences series focuses on ‘‘translational medicine’’ and cutting-edge technologies. Preclinical and clinical trials for the development of new diagostics and therapeutics as well as prevention efforts are covered whenever possible. Cameron S. Carter • Jeffrey W. Dalley Editors Brain Imaging in Behavioral Neuroscience 123 Editors Cameron S. Carter Jeffrey W. Dalley Imaging Research Center Department of Experimental Psychology Center for Neuroscience University of Cambridge University of California at Davis Downing Site Sacramento, CA 95817 Cambridge CB2 3EB USA UK ISSN 1866-3370 ISSN 1866-3389 (electronic) ISBN 978-3-642-28710-7 ISBN 978-3-642-28711-4 (eBook) DOI 10.1007/978-3-642-28711-4 Springer Heidelberg New York Dordrecht London Library of Congress Control Number: 2012938202 Ó Springer-Verlag Berlin Heidelberg 2012 This work is subject to copyright. -

A Brief Overview of Psychiatric Pharmacotherapy
A Brief Overview of Psychiatric Pharmacotherapy Joel V. Oberstar, M.D. Chief Executive Officer Disclosures • Some medications discussed are not approved by the FDA for use in the population discussed/described. • Some medications discussed are not approved by the FDA for use in the manner discussed/described. • Co-Owner: – PrairieCare and PrairieCare Medical Group – Catch LLC Disclaimer The contents of this handout are for informational purposes only and are not intended to be a substitute for professional medical advice, diagnosis, or treatment. Always seek the advice of your physician or other qualified healthcare provider with any questions you may have regarding a medical or psychiatric condition. Never disregard professional/medical advice or delay in seeking it because of something you have read in this handout. Material in this handout may be copyrighted by the author or by third parties; reasonable efforts have been made to give attribution where appropriate. Caveat Regarding the Role of Medication… Neuroscience Overview Mind Over Matter, National Institute on Drug Abuse, National Institutes of Health. Available at: http://teens.drugabuse.gov/mom/index.asp. http://medicineworld.org/images/news-blogs/brain-700997.jpg Neuroscience Overview Mind Over Matter, National Institute on Drug Abuse, National Institutes of Health. Available at: http://teens.drugabuse.gov/mom/index.asp. Neurotransmitter Receptor Source: National Institute on Drug Abuse Common Diagnoses and Associated Medications • Psychotic Disorders – Antipsychotics • Bipolar Disorders – Mood Stabilizers, Antipsychotics, & Antidepressants • Depressive Disorders – Antidepressants • Anxiety Disorders – Antidepressants & Anxiolytics • Attention Deficit Hyperactivity Disorder – Stimulants, Antidepressants, 2-Adrenergic Agents, & Strattera Classes of Medications • Anti-depressants • Stimulants and non-stimulant alternatives • Anti-psychotics (a.k.a. -

Guide for Use and Monitoring of Psychotropic Medications in Children and Adolescents
Guide for Use and Monitoring of Psychotropic Medications In Children and Adolescents This document was developed by Community Care of North Carolina with the assistance of Fostering Health NC’s Medication Management Workgroup. The information contained in this guide is not intended to substitute or act as medical advice. If you have any questions about a medication prescribed to a child or adolescent in your care, contact the prescriber or a licensed medical professional. www.ncpeds.org/fosteringhealthnc 1 | P a g e Guide for Use and Monitoring of Psychotropic Medications In Children and Adolescents (Original 3.22.17 – revised 5.22.19) Definition of Psychotropic Medication: Medication used in the treatment of mental illnesses and capable of affecting the mind, emotions, and behavior. Use of this Information: The tables below offer information for child welfare workers, care managers and foster/resource parents to use when they want to learn more about a psychotropic medication. Specific information is outlined including therapeutic class, brand and generic names, FDA approved uses, common evidence-based uses, potential side effects, and medication-specific safety/effectiveness monitoring necessary when prescribed to a child or adolescent. Because few medications have been FDA approved for use in children 5 years of age and under, there is a column in the table that lists the FDA approved status and age ranges for the approved uses of each medication. This guide also provides a color-coded quick reference guide for each medication (Appendix A), questions that a foster/resource parent should ask a prescriber (Appendix B), and a glossary of terms (Appendix C). -
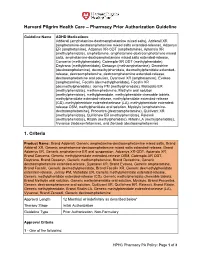
Pharmacy Prior Authorization Guideline 1. Criteria
Harvard Pilgrim Health Care – Pharmacy Prior Authorization Guideline Guideline Name ADHD Medications: Adderall (amphetamine-dextroamphetamine mixed salts), Adderall XR (amphetamine-dextroamphetamine mixed salts extended-release), Adzenys ER (amphetamine), Adzenys XR-ODT (amphetamine), Aptensio XR (methylphenidate), amphetamine, amphetamine-dextroamphetamine mixed salts, amphetamine-dextroamphetamine mixed salts extended-release, Concerta (methylphenidate), Cotempla-XR ODT (methylphenidate), Daytrana (methylphenidate), Desoxyn (methamphetamine), Dexedrine (dextroamphetamine), dexmethylphenidate, dexmethylphenidate extended- release, dextroamphetamine, dextroamphetamine extended-release, dextroamphetamine oral solution, Dyanavel XR (amphetamine), Evekeo (amphetamine), Focalin (dexmethylphenidate), Focalin XR (dexmethylphenidate), Jornay PM (methylphenidate), Metadate ER (methylphenidate), methamphetamine, Methylin oral solution (methylphenidate), methylphenidate, methylphenidate chewable tablets, methylphenidate extended-release, methylphenidate extended-release (CD), methylphenidate extended-release (LA), methylphenidate extended- release OSM, methylphenidate oral solution, Mydayis (amphetamine- dextroamphetamine), Procentra (dextroamphetamine), Quillivant XR (methylphenidate), Quillichew ER (methylphenidate), Relexxii (methylphenidate), Ritalin (methylphenidate), Ritalin LA (methylphenidate), Vyvanse (lisdexamfetamine), and Zenzedi (dextroamphetamine) 1. Criteria Product Name: Brand Adderall, Generic amphetamine-dextroamphetamine mixed -
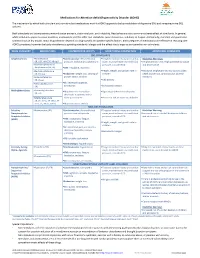
Medications for Attention Deficit/Hyperactivity Disorder (ADHD)
Medications for Attention-deficit/hyperactivity Disorder (ADHD) The mechanism by which both stimulant and non-stimulant medications work for ADHD appears to be by modulation of dopamine (DA) and norepinephrine (NE) systems. Both stimulants and atomoxetine commonly cause anorexia, abdominal pain, and irritability. Headaches are also a common adverse effect of stimulants. In general, while stimulants appear to cause insomnia, atomoxetine and the other non-stimulants cause drowsiness, sedation, or fatigue. Additionally, clonidine and guanfacine commonly cause dry mouth. Initial drug selection depends on drug-specific and patient-specific factors. Both categories of medications are effective in reducing core ADHD symptoms, however the body of evidence supporting stimulants is larger and the effect size is larger as compared to non-stimulants. DRUG CATEGORY MEDICATIONS COMMON SIDE EFFECTS MONITORING PARAMETERS ADDITIONAL COMMENTS CNS STIMULANTS Amphetamines .Amphetamine .Cardiovascular: elevated blood .Targeted cardiac history and cardiac Black Box Warnings: (IR, ODT, XR-ODT, XR-liq)* pressure, tachycardia, palpitations exam: monitor heart rate and blood .Amphetamines have a high potential for abuse .Dextroamphetamine/ pressure, consider ECG and dependence Amphetamine (IR, ER) .CNS: headache, insomnia .Dextroamphetamine .Height, weight, and growth rate in .Misuse of amphetamines may cause sudden (IR, ER, Liq) .Endocrine: weight loss, slowing of children death and serious cardiovascular adverse .Lisdexamfetamine growth rate in children reactions -

Urine Drug Toxicology and Pain Management Testing
Urine Drug Toxicology and Pain Management Testing Kara Lynch, PhD, DABCC University of California San Francisco San Francisco, CA Learning Objectives • Describe what chronic pain is, who is affected and how they are commonly treated • Explain the common methodologies used in pain management testing • Interpret urine drug testing results • Create protocols to minimize the occurrence of false positive or false negative results Pain – definition and types • Pain - an unpleasant sensory and emotional experience associated with actual or potential tissue damage • Chronic pain - pain that extends beyond the expected period of healing • Nociceptive Pain – Pain caused by tissue injury – Stimulus-evoked, high intensity – Opioid sensitive • Neuropathic Pain – Caused by nerve injury – Spontaneous activity – Develops in days or month – Opioid insensitive Chronic Pain Patients • Arthritis • Fibromyalgia – Osteoarthritis • Headaches – Rheumatoid arthritis – Migraine – Gout – Tension • Cancer – Cluster • Chronic non-cancer pain • Myofascial pain • Central pain syndrome • Neuropathic pain – CNS damage – Diabetic Peripheral – Multiple Sclerosis Neuropathy – Parkinson’s disease – Postherpetic neuralgia • Chronic abnominal pain • Neck and Back Pain – Intestinal obstructions Chronic Pain Medications • Opiates • Synthetic opioids – Codeine – Fentanyl – Morphine – Methadone • Semi-synthetic opioids – Meperidine – Oxymorphone – Tramadol – Oxycodone – Propoxyphene – Hydromorphone – Levorphanol – Hydrocodone – Tapentadol – Buprenorphine • Anticonvulsant • Muscle -

2013 National Survey on Drug Use and Health
140324 Table 1.88C Specific Hallucinogen, Inhalant, Needle, and Heroin Use in Lifetime, by Age Group: Standard Errors of Numbers in Thousands, 2012 and 2013 Total Total Aged 12-17 Aged 12-17 Aged 18-25 Aged 18-25 Aged 26+ Aged 26+ Drug or Method of Administration (2012) (2013) (2012) (2013) (2012) (2013) (2012) (2013) HALLUCINOGENS 674 707 39 35 131 133 641 678 PCP (Angel Dust, Phencyclidine) 311 346 13 8 30 30 309 342 LSD (Acid) 561 580 21 19 87 81 538 567 Peyote 338 328 10 9 31 33 331 327 Mescaline 362 378 7 6 30 35 359 375 Psilocybin (Mushrooms) 524 561 24 22 99 100 502 542 Ecstasy (MDMA) 405 464 30 24 117 114 374 442 INHALANTS 489 519 51 52 84 85 471 505 Amyl Nitrite, Poppers, Locker Room Odorizers, or Rush 311 364 21 18 40 40 306 357 Correction Fluid, Degreaser, or Cleaning Fluid 162 100 26 26 28 26 157 91 Gasoline or Lighter Fluid 184 164 31 28 40 29 174 156 Glue, Shoe Polish, or Toluene 182 175 31 31 32 30 175 168 Halothane, Ether, or Other Anesthetics 89 85 8 10 15 17 88 84 Lacquer Thinner or Other Paint Solvents 132 117 21 18 21 21 130 114 Lighter Gases (Butane, Propane) 91 80 18 15 16 17 89 77 Nitrous Oxide or Whippits 365 409 18 17 71 70 349 399 Spray Paints 123 121 34 24 28 28 115 115 Other Aerosol Sprays 154 130 24 21 33 39 149 121 NEEDLE USE1,2,3 225 265 10 10 38 40 222 261 Heroin Needle Use1,2 162 211 7 6 28 33 161 209 Cocaine Needle Use1,2 160 193 3 1 20 21 158 192 Stimulant Needle Use1,2,3 143 199 4 5 22 26 139 196 Methamphetamine Needle Use1,2,3 134 185 3 5 21 22 131 182 HEROIN 245 281 12 9 43 44 241 278 Smoke Heroin2 153 152 9 5 30 32 148 150 Sniff or Snort Heroin2 203 215 5 5 35 35 203 212 *Low precision; no estimate reported. -

World of Cognitive Enhancers
ORIGINAL RESEARCH published: 11 September 2020 doi: 10.3389/fpsyt.2020.546796 The Psychonauts’ World of Cognitive Enhancers Flavia Napoletano 1,2, Fabrizio Schifano 2*, John Martin Corkery 2, Amira Guirguis 2,3, Davide Arillotta 2,4, Caroline Zangani 2,5 and Alessandro Vento 6,7,8 1 Department of Mental Health, Homerton University Hospital, East London Foundation Trust, London, United Kingdom, 2 Psychopharmacology, Drug Misuse, and Novel Psychoactive Substances Research Unit, School of Life and Medical Sciences, University of Hertfordshire, Hatfield, United Kingdom, 3 Swansea University Medical School, Institute of Life Sciences 2, Swansea University, Swansea, United Kingdom, 4 Psychiatry Unit, Department of Clinical and Experimental Medicine, University of Catania, Catania, Italy, 5 Department of Health Sciences, University of Milan, Milan, Italy, 6 Department of Mental Health, Addictions’ Observatory (ODDPSS), Rome, Italy, 7 Department of Mental Health, Guglielmo Marconi” University, Rome, Italy, 8 Department of Mental Health, ASL Roma 2, Rome, Italy Background: There is growing availability of novel psychoactive substances (NPS), including cognitive enhancers (CEs) which can be used in the treatment of certain mental health disorders. While treating cognitive deficit symptoms in neuropsychiatric or neurodegenerative disorders using CEs might have significant benefits for patients, the increasing recreational use of these substances by healthy individuals raises many clinical, medico-legal, and ethical issues. Moreover, it has become very challenging for clinicians to Edited by: keep up-to-date with CEs currently available as comprehensive official lists do not exist. Simona Pichini, Methods: Using a web crawler (NPSfinder®), the present study aimed at assessing National Institute of Health (ISS), Italy Reviewed by: psychonaut fora/platforms to better understand the online situation regarding CEs. -

Child and Adolescent Psychopharmacology Manual
Southern Consortium for Children Athens, Hocking, Vinton 317 Board ADAMHS Board of Adams-Lawrence-Scioto Counties Gallia-Jackson-Meigs Board of ADAMHS Washington County MHAR Board CHILD AND ADOLESCENT PSYCHOPHARMACOLOGY MANUAL Issued: November 2005 Compiled by: Peg Meis, M.Ed., LPCC, Director of Crisis Services Reviewed by: Edward Lynam, M.D. & Linda Adams, R.N., MSN Designed by: Jenny Metts Preface To the User: This manual has been put together for informational purposes only. It is not meant to be used for clinical practice. Each physician will have their own methodology in treating children and adolescents, as many of these drugs have not been fully tested for use with patients under the age of 18. It is strongly advised that when questions arise regarding medication for a specific youth that those questions be referred to the treating physician. • Please be aware and attuned to the possible side effects of any medication, especially at the onset of starting any new medication. • In particular, suicidal ideation with antidepressants, stimulants, and Strattera. • Watch for adverse effects of all stimulants, such as hallucinations and psychosis. It is our hope that this manual will be helpful in the field for therapists, case managers, counselors, court personnel, and child-serving agencies. We encourage you to reproduce this manual as needed and to distribute it to all staff who may benefit from its use. This manual can also be downloaded online by going to www.scchildren.com. Southern Consortium for Children Post Office Box 956 Athens, Ohio 45701 740-593-8293 Phone / 740-592-4170 Fax Contact: Peg Meis [email protected] www.scchildren.com Table of Contents ADHD Treatment Adderal.............................................................................................................