A Multi-OMIC Characterization of Biodegradation and Microbial Community Succession Within the PET Plastisphere
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Legionella Shows a Diverse Secondary Metabolism Dependent on a Broad Spectrum Sfp-Type Phosphopantetheinyl Transferase
Legionella shows a diverse secondary metabolism dependent on a broad spectrum Sfp-type phosphopantetheinyl transferase Nicholas J. Tobias1, Tilman Ahrendt1, Ursula Schell2, Melissa Miltenberger1, Hubert Hilbi2,3 and Helge B. Bode1,4 1 Fachbereich Biowissenschaften, Merck Stiftungsprofessur fu¨r Molekulare Biotechnologie, Goethe Universita¨t, Frankfurt am Main, Germany 2 Max von Pettenkofer Institute, Ludwig-Maximilians-Universita¨tMu¨nchen, Munich, Germany 3 Institute of Medical Microbiology, University of Zu¨rich, Zu¨rich, Switzerland 4 Buchmann Institute for Molecular Life Sciences, Goethe Universita¨t, Frankfurt am Main, Germany ABSTRACT Several members of the genus Legionella cause Legionnaires’ disease, a potentially debilitating form of pneumonia. Studies frequently focus on the abundant number of virulence factors present in this genus. However, what is often overlooked is the role of secondary metabolites from Legionella. Following whole genome sequencing, we assembled and annotated the Legionella parisiensis DSM 19216 genome. Together with 14 other members of the Legionella, we performed comparative genomics and analysed the secondary metabolite potential of each strain. We found that Legionella contains a huge variety of biosynthetic gene clusters (BGCs) that are potentially making a significant number of novel natural products with undefined function. Surprisingly, only a single Sfp-like phosphopantetheinyl transferase is found in all Legionella strains analyzed that might be responsible for the activation of all carrier proteins in primary (fatty acid biosynthesis) and secondary metabolism (polyketide and non-ribosomal peptide synthesis). Using conserved active site motifs, we predict Submitted 29 June 2016 some novel compounds that are probably involved in cell-cell communication, Accepted 25 October 2016 Published 24 November 2016 differing to known communication systems. -

Genome Characteristics of a Generalist Marine Bacterial Lineage
The ISME Journal (2010), 1–15 & 2010 International Society for Microbial Ecology All rights reserved 1751-7362/10 $32.00 www.nature.com/ismej ORIGINAL ARTICLE Genome characteristics of a generalist marine bacterial lineage Ryan J Newton1, Laura E Griffin1, Kathy M Bowles1, Christof Meile1, Scott Gifford1, Carrie E Givens1, Erinn C Howard1, Eric King1, Clinton A Oakley2, Chris R Reisch3, Johanna M Rinta-Kanto1, Shalabh Sharma1, Shulei Sun1, Vanessa Varaljay3, Maria Vila-Costa1,4, Jason R Westrich5 and Mary Ann Moran1 1Department of Marine Sciences, University of Georgia, Athens, GA, USA; 2Department of Plant Biology, University of Georgia, Athens, GA, USA; 3Department of Microbiology, University of Georgia, Athens, GA, USA; 4Group of Limnology-Department of Continental Ecology, Centre d’Estudis Avanc¸ats de Blanes-CSIS, Catalunya, Spain and 5Odum School of Ecology, University of Georgia, Athens, GA, USA Members of the marine Roseobacter lineage have been characterized as ecological generalists, suggesting that there will be challenges in assigning well-delineated ecological roles and biogeochemical functions to the taxon. To address this issue, genome sequences of 32 Roseobacter isolates were analyzed for patterns in genome characteristics, gene inventory, and individual gene/ pathway distribution using three predictive frameworks: phylogenetic relatedness, lifestyle strategy and environmental origin of the isolate. For the first framework, a phylogeny containing five deeply branching clades was obtained from a concatenation of 70 conserved single-copy genes. Somewhat surprisingly, phylogenetic tree topology was not the best model for organizing genome characteristics or distribution patterns of individual genes/pathways, although it provided some predictive power. The lifestyle framework, established by grouping isolates according to evidence for heterotrophy, photoheterotrophy or autotrophy, explained more of the gene repertoire in this lineage. -

Microbial Diversity Under Extreme Euxinia: Mahoney Lake, Canada V
Geobiology (2012), 10, 223–235 DOI: 10.1111/j.1472-4669.2012.00317.x Microbial diversity under extreme euxinia: Mahoney Lake, Canada V. KLEPAC-CERAJ,1,2 C. A. HAYES,3 W. P. GILHOOLY,4 T. W. LYONS,5 R. KOLTER2 AND A. PEARSON3 1Department of Molecular Genetics, Forsyth Institute, Cambridge, MA, USA 2Department of Microbiology and Molecular Genetics, Harvard Medical School, Boston, MA, USA 3Department of Earth and Planetary Sciences, Harvard University, Cambridge, MA, USA 4Department of Earth and Planetary Sciences, Washington University, Saint Louis, MO, USA 5Department of Earth Sciences, University of California, Riverside, CA, USA ABSTRACT Mahoney Lake, British Columbia, Canada, is a stratified, 15-m deep saline lake with a euxinic (anoxic, sulfidic) hypolimnion. A dense plate of phototrophic purple sulfur bacteria is found at the chemocline, but to date the rest of the Mahoney Lake microbial ecosystem has been underexamined. In particular, the microbial community that resides in the aphotic hypolimnion and ⁄ or in the lake sediments is unknown, and it is unclear whether the sulfate reducers that supply sulfide for phototrophy live only within, or also below, the plate. Here we profiled distribu- tions of 16S rRNA genes using gene clone libraries and PhyloChip microarrays. Both approaches suggest that microbial diversity is greatest in the hypolimnion (8 m) and sediments. Diversity is lowest in the photosynthetic plate (7 m). Shallower depths (5 m, 7 m) are rich in Actinobacteria, Alphaproteobacteria, and Gammaproteo- bacteria, while deeper depths (8 m, sediments) are rich in Crenarchaeota, Natronoanaerobium, and Verrucomi- crobia. The heterogeneous distribution of Deltaproteobacteria and Epsilonproteobacteria between 7 and 8 m is consistent with metabolisms involving sulfur intermediates in the chemocline, but complete sulfate reduction in the hypolimnion. -

The Risk to Human Health from Free-Living Amoebae Interaction with Legionella in Drinking and Recycled Water Systems
THE RISK TO HUMAN HEALTH FROM FREE-LIVING AMOEBAE INTERACTION WITH LEGIONELLA IN DRINKING AND RECYCLED WATER SYSTEMS Dissertation submitted by JACQUELINE MARIE THOMAS BACHELOR OF SCIENCE (HONOURS) AND BACHELOR OF ARTS, UNSW In partial fulfillment of the requirements for the award of DOCTOR OF PHILOSOPHY in ENVIRONMENTAL ENGINEERING SCHOOL OF CIVIL AND ENVIRONMENTAL ENGINEERING FACULTY OF ENGINEERING MAY 2012 SUPERVISORS Professor Nicholas Ashbolt Office of Research and Development United States Environmental Protection Agency Cincinnati, Ohio USA and School of Civil and Environmental Engineering Faculty of Engineering The University of New South Wales Sydney, Australia Professor Richard Stuetz School of Civil and Environmental Engineering Faculty of Engineering The University of New South Wales Sydney, Australia Doctor Torsten Thomas School of Biotechnology and Biomolecular Sciences Faculty of Science The University of New South Wales Sydney, Australia ORIGINALITY STATEMENT '1 hereby declare that this submission is my own work and to the best of my knowledge it contains no materials previously published or written by another person, or substantial proportions of material which have been accepted for the award of any other degree or diploma at UNSW or any other educational institution, except where due acknowledgement is made in the thesis. Any contribution made to the research by others, with whom 1 have worked at UNSW or elsewhere, is explicitly acknowledged in the thesis. I also declare that the intellectual content of this thesis is the product of my own work, except to the extent that assistance from others in the project's design and conception or in style, presentation and linguistic expression is acknowledged.' Signed ~ ............................ -

Horizontal Operon Transfer, Plasmids, and the Evolution of Photosynthesis in Rhodobacteraceae
The ISME Journal (2018) 12:1994–2010 https://doi.org/10.1038/s41396-018-0150-9 ARTICLE Horizontal operon transfer, plasmids, and the evolution of photosynthesis in Rhodobacteraceae 1 2 3 4 1 Henner Brinkmann ● Markus Göker ● Michal Koblížek ● Irene Wagner-Döbler ● Jörn Petersen Received: 30 January 2018 / Revised: 23 April 2018 / Accepted: 26 April 2018 / Published online: 24 May 2018 © The Author(s) 2018. This article is published with open access Abstract The capacity for anoxygenic photosynthesis is scattered throughout the phylogeny of the Proteobacteria. Their photosynthesis genes are typically located in a so-called photosynthesis gene cluster (PGC). It is unclear (i) whether phototrophy is an ancestral trait that was frequently lost or (ii) whether it was acquired later by horizontal gene transfer. We investigated the evolution of phototrophy in 105 genome-sequenced Rhodobacteraceae and provide the first unequivocal evidence for the horizontal transfer of the PGC. The 33 concatenated core genes of the PGC formed a robust phylogenetic tree and the comparison with single-gene trees demonstrated the dominance of joint evolution. The PGC tree is, however, largely incongruent with the species tree and at least seven transfers of the PGC are required to reconcile both phylogenies. 1234567890();,: 1234567890();,: The origin of a derived branch containing the PGC of the model organism Rhodobacter capsulatus correlates with a diagnostic gene replacement of pufC by pufX. The PGC is located on plasmids in six of the analyzed genomes and its DnaA- like replication module was discovered at a conserved central position of the PGC. A scenario of plasmid-borne horizontal transfer of the PGC and its reintegration into the chromosome could explain the current distribution of phototrophy in Rhodobacteraceae. -
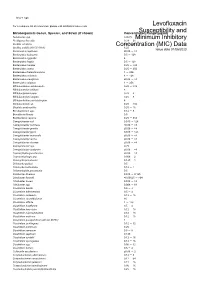
Susceptibility and Resistance Data
toku-e logo For a complete list of references, please visit antibiotics.toku-e.com Levofloxacin Microorganism Genus, Species, and Strain (if shown) Concentration Range (μg/ml)Susceptibility and Aeromonas spp. 0.0625 Minimum Inhibitory Alcaligenes faecalis 0.39 - 25 Bacillus circulans Concentration0.25 - 8 (MIC) Data Bacillus subtilis (ATCC 6051) 6.25 Issue date 01/06/2020 Bacteroides capillosus ≤0.06 - >8 Bacteroides distasonis 0.5 - 128 Bacteroides eggerthii 4 Bacteroides fragilis 0.5 - 128 Bacteroides merdae 0.25 - >32 Bacteroides ovatus 0.25 - 256 Bacteroides thetaiotaomicron 1 - 256 Bacteroides uniformis 4 - 128 Bacteroides ureolyticus ≤0.06 - >8 Bacteroides vulgatus 1 - 256 Bifidobacterium adolescentis 0.25 - >32 Bifidobacterium bifidum 8 Bifidobacterium breve 0.25 - 8 Bifidobacterium longum 0.25 - 8 Bifidobacterium pseudolongum 8 Bifidobacterium sp. 0.25 - >32 Bilophila wadsworthia 0.25 - 16 Brevibacterium spp. 0.12 - 8 Brucella melitensis 0.5 Burkholderia cepacia 0.25 - 512 Campylobacter coli 0.015 - 128 Campylobacter concisus ≤0.06 - >8 Campylobacter gracilis ≤0.06 - >8 Campylobacter jejuni 0.015 - 128 Campylobacter mucosalis ≤0.06 - >8 Campylobacter rectus ≤0.06 - >8 Campylobacter showae ≤0.06 - >8 Campylobacter spp. 0.25 Campylobacter sputorum ≤0.06 - >8 Capnocytophaga ochracea ≤0.06 - >8 Capnocytophaga spp. 0.006 - 2 Chlamydia pneumonia 0.125 - 1 Chlamydia psittaci 0.5 Chlamydia trachomatis 0.12 - 1 Chlamydophila pneumonia 0.5 Citrobacter diversus 0.015 - 0.125 Citrobacter freundii ≤0.00625 - >64 Citrobacter koseri 0.015 - -

Aquascreen® Legionella Species Qpcr Detection Kit
AquaScreen® Legionella species qPCR Detection Kit INSTRUCTIONS FOR USE FOR USE IN RESEARCH AND QUALITY CONTROL Symbols Lot No. Cat. No. Expiry date Storage temperature Number of reactions Manufacturer INDICATION The AquaScreen® Legionella species qPCR Detection kit is specifically designed for the quantitative detection of several Legionella species in water samples prepared with the AquaScreen® FastExt- ract kit. Its design complies with the requirements of AFNOR T90-471 and ISO/TS 12869:2012. Legionella are ubiquitous bacteria in surface water and moist soil, where they parasitize protozoa. The optimal growth temperature lies between +15 and +45 °C, whereas these gram-negative bacteria are dormant below 20 °C and do not survive above 60 °C. Importantly, Legionella are well-known as opportunistic intracellular human pathogens causing Legionnaires’ disease and Pontiac fever. The transmission occurs through inhalation of contami- nated aerosols generated by an infected source (e.g. human-made water systems like shower- heads, sink faucets, heaters, cooling towers, and many more). In order to efficiently prevent Legionella outbreaks, water safety control measures need syste- matic application but also reliable validation by fast Legionella testing. TEST PRINCIPLE The AquaScreen® Legionella species Kit uses qPCR for quantitative detection of legionella in wa- ter samples. In contrast to more time-consuming culture-based methods, AquaScreen® assays need less than six hours including sample preparation and qPCR to reliably detect Legionella. Moreover, the AquaScreen® qPCR assay has proven excellent performance in terms of specificity and sensitivity: other bacterial genera remain undetected whereas linear quantification is obtai- ned up to 1 x 106 particles per sample, therefore requiring no material dilution. -

Genomic Analyses of Two Alteromonas Stellipolaris Strains Reveal Traits
www.nature.com/scientificreports OPEN Genomic analyses of two Alteromonas stellipolaris strains reveal traits with potential Received: 7 August 2018 Accepted: 27 November 2018 biotechnological applications Published: xx xx xxxx Marta Torres1,2,3, Kar-Wai Hong4, Teik-Min Chong4, José Carlos Reina1, Kok-Gan Chan 4,5, Yves Dessaux3 & Inmaculada Llamas 1,2 The Alteromonas stellipolaris strains PQQ-42 and PQQ-44, previously isolated from a fsh hatchery, have been selected on the basis of their strong quorum quenching (QQ) activity, as well as their ability to reduce Vibrio-induced mortality on the coral Oculina patagonica. In this study, the genome sequences of both strains were determined and analyzed in order to identify the mechanism responsible for QQ activity. Both PQQ-42 and PQQ-44 were found to degrade a wide range of N-acylhomoserine lactone (AHL) QS signals, possibly due to the presence of an aac gene which encodes an AHL amidohydrolase. In addition, the diferent colony morphologies exhibited by the strains could be related to the diferences observed in genes encoding cell wall biosynthesis and exopolysaccharide (EPS) production. The PQQ-42 strain produces more EPS (0.36 g l−1) than the PQQ-44 strain (0.15 g l−1), whose chemical compositions also difer. Remarkably, PQQ-44 EPS contains large amounts of fucose, a sugar used in high-value biotechnological applications. Furthermore, the genome of strain PQQ-42 contained a large non- ribosomal peptide synthase (NRPS) cluster with a previously unknown genetic structure. The synthesis of enzymes and other bioactive compounds were also identifed, indicating that PQQ-42 and PQQ-44 could have biotechnological applications. -

Immunoproteomic Identification of Biomarkers for Diagnosis of Legionellosis
Immunoproteomic identification of biomarkers for diagnosis of legionellosis Submitted in total fulfilment of the requirements for the degree of Doctor of philosophy by Kaylass Poorun Department of Chemistry and Biotechnology Faculty of Science, Engineering and Technology Swinburne University of Technology Australia 2014 Abstract Abstract Legionellosis, a disease with significant mortality and morbidity rates, is considered to be the second most frequent cause of severe community-acquired pneumonia. It is difficult to distinguish from other types of pneumonia due to similar clinical manifestations. Several studies have demonstrated the inadequacies of current diagnostic tests for confirming Legionella infections. This study was aimed at identifying biomarkers that can be used in an improved test. A comparative proteomic analysis, using DIGE, was carried out between L. pneumophila ATCC33152 and L. longbeachae NSW150 and D4968 isolates. While many homologous proteins were found to be commonly expressed, numerous others were identified to be differentially expressed under similar in vitro conditions suggesting that the two species have different lifestyles and infection strategies. The bacterial immunoglobulin domain containing protein, found to share sequence homology to Type V secretion proteins intimin and invasin, is not known to be present in Legionella. Human sera containing antibodies against Legionella from a set of blind samples were identified by ELISA. Downstream analyses revealed that diverse immunogens may be responsible for eliciting immune response in different Legionella species which in turn show little to no congeneric cross-reactivity. To the best of our knowledge, this is a unique finding not previously reported. Several serological diagnostic tests currently in use do not include many Legionella species in their testing panel, which may be a reason for many Legionella species being under-reported. -

Mechanisms Driving Genome Reduction of a Novel Roseobacter Lineage Showing
bioRxiv preprint doi: https://doi.org/10.1101/2021.01.15.426902; this version posted January 16, 2021. The copyright holder for this preprint (which was not certified by peer review) is the author/funder, who has granted bioRxiv a license to display the preprint in perpetuity. It is made available under aCC-BY-NC-ND 4.0 International license. 1 Mechanisms Driving Genome Reduction of a Novel Roseobacter Lineage Showing 2 Vitamin B12 Auxotrophy 3 4 Xiaoyuan Feng1, Xiao Chu1, Yang Qian1, Michael W. Henson2a, V. Celeste Lanclos2, Fang 5 Qin3, Yanlin Zhao3, J. Cameron Thrash2, Haiwei Luo1* 6 7 1Simon F. S. Li Marine Science Laboratory, School of Life Sciences and State Key 8 Laboratory of Agrobiotechnology, The Chinese University of Hong Kong, Shatin, Hong 9 Kong SAR 10 2Department of Biological Sciences, University of Southern California, Los Angeles, CA 11 USA 12 3Fujian Provincial Key Laboratory of Agroecological Processing and Safety Monitoring, 13 College of Life Sciences, Fujian Agriculture and Forestry University, Fuzhou, Fujian, China 14 a Current Affiliation: Department of Geophysical Sciences, University of Chicago, Chicago, 15 Illinois, USA 16 17 *Corresponding author: 18 Haiwei Luo 19 School of Life Sciences, The Chinese University of Hong Kong 20 Shatin, Hong Kong SAR 21 Phone: (+852) 3943-6121 22 Fax: (+852) 2603-5646 23 E-mail: [email protected] 24 25 Keywords: Roseobacter, CHUG, genome reduction, vitamin B12 auxotrophy 26 27 bioRxiv preprint doi: https://doi.org/10.1101/2021.01.15.426902; this version posted January 16, 2021. The copyright holder for this preprint (which was not certified by peer review) is the author/funder, who has granted bioRxiv a license to display the preprint in perpetuity. -

Photosynthesis Is Widely Distributed Among Proteobacteria As Demonstrated by the Phylogeny of Puflm Reaction Center Proteins
fmicb-08-02679 January 20, 2018 Time: 16:46 # 1 ORIGINAL RESEARCH published: 23 January 2018 doi: 10.3389/fmicb.2017.02679 Photosynthesis Is Widely Distributed among Proteobacteria as Demonstrated by the Phylogeny of PufLM Reaction Center Proteins Johannes F. Imhoff1*, Tanja Rahn1, Sven Künzel2 and Sven C. Neulinger3 1 Research Unit Marine Microbiology, GEOMAR Helmholtz Centre for Ocean Research, Kiel, Germany, 2 Max Planck Institute for Evolutionary Biology, Plön, Germany, 3 omics2view.consulting GbR, Kiel, Germany Two different photosystems for performing bacteriochlorophyll-mediated photosynthetic energy conversion are employed in different bacterial phyla. Those bacteria employing a photosystem II type of photosynthetic apparatus include the phototrophic purple bacteria (Proteobacteria), Gemmatimonas and Chloroflexus with their photosynthetic relatives. The proteins of the photosynthetic reaction center PufL and PufM are essential components and are common to all bacteria with a type-II photosynthetic apparatus, including the anaerobic as well as the aerobic phototrophic Proteobacteria. Edited by: Therefore, PufL and PufM proteins and their genes are perfect tools to evaluate the Marina G. Kalyuzhanaya, phylogeny of the photosynthetic apparatus and to study the diversity of the bacteria San Diego State University, United States employing this photosystem in nature. Almost complete pufLM gene sequences and Reviewed by: the derived protein sequences from 152 type strains and 45 additional strains of Nikolai Ravin, phototrophic Proteobacteria employing photosystem II were compared. The results Research Center for Biotechnology (RAS), Russia give interesting and comprehensive insights into the phylogeny of the photosynthetic Ivan A. Berg, apparatus and clearly define Chromatiales, Rhodobacterales, Sphingomonadales as Universität Münster, Germany major groups distinct from other Alphaproteobacteria, from Betaproteobacteria and from *Correspondence: Caulobacterales (Brevundimonas subvibrioides). -

Microbial and Mineralogical Characterizations of Soils Collected from the Deep Biosphere of the Former Homestake Gold Mine, South Dakota
University of Nebraska - Lincoln DigitalCommons@University of Nebraska - Lincoln US Department of Energy Publications U.S. Department of Energy 2010 Microbial and Mineralogical Characterizations of Soils Collected from the Deep Biosphere of the Former Homestake Gold Mine, South Dakota Gurdeep Rastogi South Dakota School of Mines and Technology Shariff Osman Lawrence Berkeley National Laboratory Ravi K. Kukkadapu Pacific Northwest National Laboratory, [email protected] Mark Engelhard Pacific Northwest National Laboratory Parag A. Vaishampayan California Institute of Technology See next page for additional authors Follow this and additional works at: https://digitalcommons.unl.edu/usdoepub Part of the Bioresource and Agricultural Engineering Commons Rastogi, Gurdeep; Osman, Shariff; Kukkadapu, Ravi K.; Engelhard, Mark; Vaishampayan, Parag A.; Andersen, Gary L.; and Sani, Rajesh K., "Microbial and Mineralogical Characterizations of Soils Collected from the Deep Biosphere of the Former Homestake Gold Mine, South Dakota" (2010). US Department of Energy Publications. 170. https://digitalcommons.unl.edu/usdoepub/170 This Article is brought to you for free and open access by the U.S. Department of Energy at DigitalCommons@University of Nebraska - Lincoln. It has been accepted for inclusion in US Department of Energy Publications by an authorized administrator of DigitalCommons@University of Nebraska - Lincoln. Authors Gurdeep Rastogi, Shariff Osman, Ravi K. Kukkadapu, Mark Engelhard, Parag A. Vaishampayan, Gary L. Andersen, and Rajesh K. Sani This article is available at DigitalCommons@University of Nebraska - Lincoln: https://digitalcommons.unl.edu/ usdoepub/170 Microb Ecol (2010) 60:539–550 DOI 10.1007/s00248-010-9657-y SOIL MICROBIOLOGY Microbial and Mineralogical Characterizations of Soils Collected from the Deep Biosphere of the Former Homestake Gold Mine, South Dakota Gurdeep Rastogi & Shariff Osman & Ravi Kukkadapu & Mark Engelhard & Parag A.