Long Non‐Coding RNA Uc.291 Controls Epithelial Differentiation By
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

ACTL6A Promotes the Proliferation of Esophageal Squamous Cell Carcinoma Cells and Correlates with Poor Clinical Outcomes
OncoTargets and Therapy Dovepress open access to scientific and medical research Open Access Full Text Article ORIGINAL RESEARCH ACTL6A Promotes the Proliferation of Esophageal Squamous Cell Carcinoma Cells and Correlates with Poor Clinical Outcomes This article was published in the following Dove Press journal: OncoTargets and Therapy Rui-zhe Li1 Background: ACTL6A, a regulatory subunit of ATP-dependent chromatin-remodeling Yun-yun Li1,2 complexes SWI/SNF, has been identified as a central oncogenic driver in many tumor types. Hui Qin1 Materials and Methods: We used immunohistochemistry (IHC) to detect ACTL6A Shan-shan Li1 expression in esophageal squamous cell carcinoma (ESCC) tissues. Then, the effect of ACTL6A on proliferation and DNA synthesis was explored by using cell counting kit 8 1 Department of Pathology, School of (CCK8) and EdU retention assays. The potential oncogenic mechanism of ACTL6A in Basic Medical Sciences, Zhengzhou University and First Affiliated Hospital of ESCC cells was also analyzed by flow cytometry and Western blotting. We further estab Zhengzhou University, Zhengzhou, lished an ESCC xenograft mouse model to validate the in vitro results. Henan 450000, People’s Republic of China; 2Department of Stomatology, First Results: ACTL6A expression, localized in cancer cell nuclei, was markedly higher in ESCC Affiliated Hospital of Zhengzhou tissues than in the corresponding noncancerous tissues (P<0.001) and was positively asso University, Zhengzhou, Henan 450000, ciated with tumor size, histological differentiation, T stage and tumor-node-metastasis People’s Republic of China (TNM) stage. Kaplan–Meier analysis revealed that high ACTL6A expression was signifi cantly associated with poor overall survival (OS) (P = 0.008, HR= 2.562, 95% CI: 1.241– 5.289), and decision curve analysis (DCA) demonstrated that ACTL6A could increase the clinical prognostic efficiency of the original clinical prediction model. -

Molecular and Physiological Basis for Hair Loss in Near Naked Hairless and Oak Ridge Rhino-Like Mouse Models: Tracking the Role of the Hairless Gene
University of Tennessee, Knoxville TRACE: Tennessee Research and Creative Exchange Doctoral Dissertations Graduate School 5-2006 Molecular and Physiological Basis for Hair Loss in Near Naked Hairless and Oak Ridge Rhino-like Mouse Models: Tracking the Role of the Hairless Gene Yutao Liu University of Tennessee - Knoxville Follow this and additional works at: https://trace.tennessee.edu/utk_graddiss Part of the Life Sciences Commons Recommended Citation Liu, Yutao, "Molecular and Physiological Basis for Hair Loss in Near Naked Hairless and Oak Ridge Rhino- like Mouse Models: Tracking the Role of the Hairless Gene. " PhD diss., University of Tennessee, 2006. https://trace.tennessee.edu/utk_graddiss/1824 This Dissertation is brought to you for free and open access by the Graduate School at TRACE: Tennessee Research and Creative Exchange. It has been accepted for inclusion in Doctoral Dissertations by an authorized administrator of TRACE: Tennessee Research and Creative Exchange. For more information, please contact [email protected]. To the Graduate Council: I am submitting herewith a dissertation written by Yutao Liu entitled "Molecular and Physiological Basis for Hair Loss in Near Naked Hairless and Oak Ridge Rhino-like Mouse Models: Tracking the Role of the Hairless Gene." I have examined the final electronic copy of this dissertation for form and content and recommend that it be accepted in partial fulfillment of the requirements for the degree of Doctor of Philosophy, with a major in Life Sciences. Brynn H. Voy, Major Professor We have read this dissertation and recommend its acceptance: Naima Moustaid-Moussa, Yisong Wang, Rogert Hettich Accepted for the Council: Carolyn R. -

Increased ACTL6A Occupancy Within Mswi/SNF Chromatin Remodelers
bioRxiv preprint doi: https://doi.org/10.1101/2021.03.22.435873; this version posted March 22, 2021. The copyright holder for this preprint (which was not certified by peer review) is the author/funder. All rights reserved. No reuse allowed without permission. Increased ACTL6A Occupancy Within mSWI/SNF Chromatin Remodelers Drives Human Squamous Cell Carcinoma Chiung-Ying Chang1,2,7, Zohar Shipony3,7, Ann Kuo1,2, Kyle M. Loh4,5, William J. Greenleaf3,6, Gerald R. Crabtree1,2,5* 1Howard Hughes Medical Institute, Stanford University School of Medicine, Stanford, California 94305, USA. 2Department of Pathology, Stanford University School of Medicine, Stanford, California 94305, USA. 3Department of Genetics, Stanford University School of Medicine, Stanford, California 94305, USA. 4Institute for Stem Cell Biology and Regenerative Medicine, Stanford University School of Medicine, Stanford, California 94305, USA. 5Department of Developmental Biology, Stanford University School of Medicine, Stanford, California 94305, USA. 6Department of Applied Physics, Stanford University, Stanford, California 94305, USA. 7These authors contributed equally. *Corresponding author: Gerald R. Crabtree, [email protected] Summary Mammalian SWI/SNF (BAF) chromatin remodelers play dosage-sensitive roles in many human malignancies and neurologic disorders. The gene encoding the BAF subunit, ACTL6A, is amplified at an early stage in the development of squamous cell carcinomas (SCCs), but its oncogenic role remains unclear. Here we demonstrate that ACTL6A overexpression leads to its stoichiometric assembly into BAF complexes and drives its interaction and engagement with specific regulatory regions in the genome. In normal epithelial cells, ACTL6A was sub-stoichiometric to other BAF subunits. However, increased ACTL6A levels by ectopic expression or in SCC cells led to near-saturation of ACTL6A within BAF complexes. -

Bioinformatics Analyses of Genomic Imprinting
Bioinformatics Analyses of Genomic Imprinting Dissertation zur Erlangung des Grades des Doktors der Naturwissenschaften der Naturwissenschaftlich-Technischen Fakultät III Chemie, Pharmazie, Bio- und Werkstoffwissenschaften der Universität des Saarlandes von Barbara Hutter Saarbrücken 2009 Tag des Kolloquiums: 08.12.2009 Dekan: Prof. Dr.-Ing. Stefan Diebels Berichterstatter: Prof. Dr. Volkhard Helms Priv.-Doz. Dr. Martina Paulsen Vorsitz: Prof. Dr. Jörn Walter Akad. Mitarbeiter: Dr. Tihamér Geyer Table of contents Summary________________________________________________________________ I Zusammenfassung ________________________________________________________ I Acknowledgements _______________________________________________________II Abbreviations ___________________________________________________________ III Chapter 1 – Introduction __________________________________________________ 1 1.1 Important terms and concepts related to genomic imprinting __________________________ 2 1.2 CpG islands as regulatory elements ______________________________________________ 3 1.3 Differentially methylated regions and imprinting clusters_____________________________ 6 1.4 Reading the imprint __________________________________________________________ 8 1.5 Chromatin marks at imprinted regions___________________________________________ 10 1.6 Roles of repetitive elements ___________________________________________________ 12 1.7 Functional implications of imprinted genes _______________________________________ 14 1.8 Evolution and parental conflict ________________________________________________ -

Expressed Gene Fusions As Frequent Drivers of Poor Outcomes in Hormone Receptor–Positive Breast Cancer
Published OnlineFirst December 14, 2017; DOI: 10.1158/2159-8290.CD-17-0535 RESEARCH ARTICLE Expressed Gene Fusions as Frequent Drivers of Poor Outcomes in Hormone Receptor–Positive Breast Cancer Karina J. Matissek1,2, Maristela L. Onozato3, Sheng Sun1,2, Zongli Zheng2,3,4, Andrew Schultz1, Jesse Lee3, Kristofer Patel1, Piiha-Lotta Jerevall2,3, Srinivas Vinod Saladi1,2, Allison Macleay3, Mehrad Tavallai1,2, Tanja Badovinac-Crnjevic5, Carlos Barrios6, Nuran Beşe7, Arlene Chan8, Yanin Chavarri-Guerra9, Marcio Debiasi6, Elif Demirdögen10, Ünal Egeli10, Sahsuvar Gökgöz10, Henry Gomez11, Pedro Liedke6, Ismet Tasdelen10, Sahsine Tolunay10, Gustavo Werutsky6, Jessica St. Louis1, Nora Horick12, Dianne M. Finkelstein2,12, Long Phi Le2,3, Aditya Bardia1,2, Paul E. Goss1,2, Dennis C. Sgroi2,3, A. John Iafrate2,3, and Leif W. Ellisen1,2 ABSTRACT We sought to uncover genetic drivers of hormone receptor–positive (HR+) breast cancer, using a targeted next-generation sequencing approach for detecting expressed gene rearrangements without prior knowledge of the fusion partners. We identified inter- genic fusions involving driver genes, including PIK3CA, AKT3, RAF1, and ESR1, in 14% (24/173) of unselected patients with advanced HR+ breast cancer. FISH confirmed the corresponding chromo- somal rearrangements in both primary and metastatic tumors. Expression of novel kinase fusions in nontransformed cells deregulates phosphoprotein signaling, cell proliferation, and survival in three- dimensional culture, whereas expression in HR+ breast cancer models modulates estrogen-dependent growth and confers hormonal therapy resistance in vitro and in vivo. Strikingly, shorter overall survival was observed in patients with rearrangement-positive versus rearrangement-negative tumors. Cor- respondingly, fusions were uncommon (<5%) among 300 patients presenting with primary HR+ breast cancer. -

Inactivation of the PBRM1 Tumor Suppressor Gene Amplifies
Inactivation of the PBRM1 tumor suppressor gene − − amplifies the HIF-response in VHL / clear cell renal carcinoma Wenhua Gaoa, Wei Lib,c, Tengfei Xiaoa,b,c, Xiaole Shirley Liub,c, and William G. Kaelin Jr.a,d,1 aDepartment of Medical Oncology, Dana-Farber Cancer Institute and Brigham and Women’s Hospital, Harvard Medical School, Boston, MA 02115; bCenter for Functional Cancer Epigenetics, Dana-Farber Cancer Institute, Boston, MA 02215; cDepartment of Biostatistics and Computational Biology, Dana-Farber Cancer Institute and Harvard T.H. Chan School of Public Health, Boston, MA 02115; and dHoward Hughes Medical Institute, Chevy Chase, MD 20815 Contributed by William G. Kaelin, Jr., December 1, 2016 (sent for review October 31, 2016; reviewed by Charles W. M. Roberts and Ali Shilatifard) Most clear cell renal carcinomas (ccRCCs) are initiated by somatic monolayer culture and in soft agar (10). These effects were not, inactivation of the VHL tumor suppressor gene. The VHL gene prod- however, proven to be on-target, and were not interrogated in uct, pVHL, is the substrate recognition unit of an ubiquitin ligase vivo. As a step toward understanding the role of BAF180 in that targets the HIF transcription factor for proteasomal degrada- ccRCC, we asked whether BAF180 participates in the canonical tion; inappropriate expression of HIF target genes drives renal car- PBAF complex in ccRCC cell lines and whether loss of BAF180 cinogenesis. Loss of pVHL is not sufficient, however, to cause ccRCC. measurably alters ccRCC behavior in cell culture and in mice. Additional cooperating genetic events, including intragenic muta- tions and copy number alterations, are required. -

NIH Public Access Author Manuscript J Hepatol
NIH Public Access Author Manuscript J Hepatol. Author manuscript; available in PMC 2008 April 1. NIH-PA Author ManuscriptPublished NIH-PA Author Manuscript in final edited NIH-PA Author Manuscript form as: J Hepatol. 2008 February ; 48(2): 276±288. Small Proline Rich Proteins (SPRR) Function as SH3 Domain Ligands, Increase Resistance to Injury and are Associated with Epithelial-Mesenchymal Transition (EMT) in cholangiocytes Anthony J. Demetris, M.D.1,2,*, Susan Specht, M.S.1,2, Isao Nozaki, M.D.1,2,+, John G. Lunz III1,2,3, Donna Beer Stolz, PhD.4, Noriko Murase, M.D.1,3, and Tong Wu, M.D., PhD.2 1Thomas E Starzl Transplantation Institute, University of Pittsburgh Medical Center, Pittsburgh, PA 15213 2Department of Pathology, Division of Transplantation, University of Pittsburgh Medical Center, Pittsburgh, PA 15213 3Department of Surgery, Division of Transplantation, University of Pittsburgh Medical Center, Pittsburgh, PA 15213 4Department of Cell Biology and Physiology, University of Pittsburgh School of Medicine, Pittsburgh, Pennsylvania 15261 +Isao Nozaki, M.D., National Shikoku Cancer Center Hospital, Department of Surgery, 13 Horinouchi, Matsuyama, JAPAN 790-0007 Abstract Background/Aims—Deficient biliary epithelial cell (BEC) expression of small proline rich protein (SPRR) 2A in IL-6-/- mice is associated with defective biliary barrier function after bile duct ligation. And numerous gene array expression studies show SPRR2A to commonly be among the most highly upregulated genes in many non-squamous, stressed and remodeling barrier epithelia. Since the function of SPRR in these circumstances is unknown, we tested the exploratory hypothesis that BEC SPRR2A expression contributes to BEC barrier function and wound repair. -

Rats and Axolotls Share a Common Molecular Signature After Spinal Cord Injury Enriched in Collagen-1
Rats and axolotls share a common molecular signature after spinal cord injury enriched in collagen-1 Athanasios Didangelos1, Katalin Bartus1, Jure Tica1, Bernd Roschitzki2, Elizabeth J. Bradbury1 1Wolfson CARD King’s College London, United Kingdom. 2Centre for functional Genomics, ETH Zurich, Switzerland. Running title: spinal cord injury in rats and axolotls Correspondence: A Didangelos: [email protected] SUPPLEMENTAL FIGURES AND LEGENDS Supplemental Fig. 1: Rat 7 days microarray differentially regulated transcripts. A-B: Protein-protein interaction networks of upregulated (A) and downregulated (B) transcripts identified by microarray gene expression profiling of rat SCI (4 sham versus 4 injured spinal cord samples) 7 days post-injury. Microarray expression data and experimental information is publicly available online (https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE45006) and is also summarised in Supplemental Table 1. Protein-protein interaction networks were performed in StringDB using the full range of protein interaction scores (0.15 – 0.99) to capture maximum evidence of proteins’ interactions. Networks were then further analysed for betweeness centrality and gene ontology (GO) annotations (BinGO) in Cytoscape. Node colour indicates betweeness centrality while edge colour and thickness indicate interaction score based on predicted functional links between nodes (green: low values; red: high values). C-D: The top 10 upregulated (C) or downregulated (D) transcripts sorted by betweeness centrality score in protein-protein interaction networks shown in A & B. E-F: Biological process GO analysis was performed on networks of upregulated and downregulated genes using BinGO in Cytoscape. Graphs indicate the 20 most significant GO categories and the number of genes in each category. -

Organizing Knowledge to Enable Faster Data Interpretation in COVID
F1000Research 2021, 10(ELIXIR):703 Last updated: 01 SEP 2021 DATA NOTE Organizing knowledge to enable faster data interpretation in COVID-19 research [version 1; peer review: 2 approved with reservations] Joseph Hearnshaw, Marco Brandizi, Ajit Singh , Chris Rawlings , Keywan Hassani-Pak Rothamsted Research, Harpenden, AL5 2JQ, UK v1 First published: 30 Jul 2021, 10(ELIXIR):703 Open Peer Review https://doi.org/10.12688/f1000research.54071.1 Latest published: 30 Jul 2021, 10(ELIXIR):703 https://doi.org/10.12688/f1000research.54071.1 Reviewer Status Invited Reviewers Abstract Enormous volumes of COVID-19 research data have been published 1 2 and this continues to increase daily. This creates challenges for researchers to interpret, prioritize and summarize their own findings version 1 in the context of published literature, clinical trials, and a multitude of 30 Jul 2021 report report databases. Overcoming the data interpretation bottleneck is vital to help researchers to be more efficient in their quest to identify COVID- 1. Justin Reese , Lawrence Berkeley National 19 risk factors, potential treatments, drug side-effects, and much more. As a proof of concept, we have organized and integrated a Laboratory, Berkeley, USA range of COVID-19 and human biomedical data and literature into a knowledge graph (KG). Here we present the datasets we have 2. Keith Hall , Google Research, New York, integrated so far and the content of the KG which consists of 674,969 USA biological concepts and over 1.6 million relationships between them. The COVID-19 KG is available via KnetMiner, an interactive online Any reports and responses or comments on the platform for gene discovery and knowledge mining, or via RDF and article can be found at the end of the article. -

Supp Table 6.Pdf
Supplementary Table 6. Processes associated to the 2037 SCL candidate target genes ID Symbol Entrez Gene Name Process NM_178114 AMIGO2 adhesion molecule with Ig-like domain 2 adhesion NM_033474 ARVCF armadillo repeat gene deletes in velocardiofacial syndrome adhesion NM_027060 BTBD9 BTB (POZ) domain containing 9 adhesion NM_001039149 CD226 CD226 molecule adhesion NM_010581 CD47 CD47 molecule adhesion NM_023370 CDH23 cadherin-like 23 adhesion NM_207298 CERCAM cerebral endothelial cell adhesion molecule adhesion NM_021719 CLDN15 claudin 15 adhesion NM_009902 CLDN3 claudin 3 adhesion NM_008779 CNTN3 contactin 3 (plasmacytoma associated) adhesion NM_015734 COL5A1 collagen, type V, alpha 1 adhesion NM_007803 CTTN cortactin adhesion NM_009142 CX3CL1 chemokine (C-X3-C motif) ligand 1 adhesion NM_031174 DSCAM Down syndrome cell adhesion molecule adhesion NM_145158 EMILIN2 elastin microfibril interfacer 2 adhesion NM_001081286 FAT1 FAT tumor suppressor homolog 1 (Drosophila) adhesion NM_001080814 FAT3 FAT tumor suppressor homolog 3 (Drosophila) adhesion NM_153795 FERMT3 fermitin family homolog 3 (Drosophila) adhesion NM_010494 ICAM2 intercellular adhesion molecule 2 adhesion NM_023892 ICAM4 (includes EG:3386) intercellular adhesion molecule 4 (Landsteiner-Wiener blood group)adhesion NM_001001979 MEGF10 multiple EGF-like-domains 10 adhesion NM_172522 MEGF11 multiple EGF-like-domains 11 adhesion NM_010739 MUC13 mucin 13, cell surface associated adhesion NM_013610 NINJ1 ninjurin 1 adhesion NM_016718 NINJ2 ninjurin 2 adhesion NM_172932 NLGN3 neuroligin -
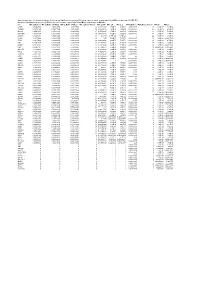
Supplementary Table 6. Summary of Mutation Frequency And
Supplementary table 6. Summary of mutation frequency and significance per microsatellite and per gene in exome sequencing of 24 and MiSeq sequencing of 93 MSI CRCs Summary of mutation frequency and significance per gene in exome sequencing of 24 and MiSeq sequencing of 93 MSI CRCs Gene MiSeq_MutFreq MiSeq_MutFreqCI95High MiSeq_MutFreqCI95Low MiSeq_MutatedTumors MiSeq_NAF MiSeq_P MiSeq_Q WXS_MutFreq WXS_MutatedTumors WXS_P WXS_Q CASP5 0.989247312 1.006838564 0.97165606 92 0.666666667 1.70E-89 4.83E-88 0.791666667 19 2.12E-17 1.33E-14 TGFBR2 0.967741935 0.997877874 0.937605997 90 0.883484163 1.48E-96 6.99E-95 0.833333333 20 4.39E-19 4.13E-16 SLC9A8 0.935483871 0.977386193 0.893581549 87 0.655034325 1.70E-89 4.83E-88 0.458333333 11 5.71E-07 5.25E-05 MIS18BP1 0.913978495 0.961803749 0.86615324 85 0.488888889 7.02E-61 9.07E-60 0.708333333 17 1.51E-08 1.97E-06 ACVR2A 0.892473118 0.945310572 0.839635665 83 0.80625 4.77E-128 6.77E-126 1 24 5.63E-36 2.12E-32 CDH26 0.88172043 0.936801974 0.826638886 82 0.55 4.12E-79 7.31E-78 0.416666667 10 5.46E-06 0.000331959 TFAM 0.88172043 0.936801974 0.826638886 82 0.620192308 4.12E-79 7.31E-78 0.583333333 14 2.52E-10 4.75E-08 ATR 0.860215054 0.919360254 0.801069854 80 0.632996633 2.72E-75 4.30E-74 0.583333333 14 3.89E-09 5.86E-07 AASDH 0.838709677 0.901442635 0.77597672 78 0.832712022 1.27E-71 1.80E-70 0.416666667 10 5.46E-06 0.000331959 USF3 0.806451613 0.873837619 0.739065607 75 0.771428571 5.39E-50 4.25E-49 0.5 12 0.002548628 0.01890901 SLC35F5 0.76344086 0.83592506 0.69095666 71 0.607908352 8.90E-60 1.05E-58 -

Residual Complexes Containing SMARCA2 (BRM) Underlie the Oncogenic Drive of SMARCA4 (BRG1) Mutation
Residual Complexes Containing SMARCA2 (BRM) Underlie the Oncogenic Drive of SMARCA4 (BRG1) Mutation The Harvard community has made this article openly available. Please share how this access benefits you. Your story matters Citation Wilson, B. G., K. C. Helming, X. Wang, Y. Kim, F. Vazquez, Z. Jagani, W. C. Hahn, and C. W. M. Roberts. 2014. “Residual Complexes Containing SMARCA2 (BRM) Underlie the Oncogenic Drive of SMARCA4 (BRG1) Mutation.” Molecular and Cellular Biology 34 (6): 1136–44. doi:10.1128/MCB.01372-13. Citable link http://nrs.harvard.edu/urn-3:HUL.InstRepos:41542741 Terms of Use This article was downloaded from Harvard University’s DASH repository, and is made available under the terms and conditions applicable to Other Posted Material, as set forth at http:// nrs.harvard.edu/urn-3:HUL.InstRepos:dash.current.terms-of- use#LAA Residual Complexes Containing SMARCA2 (BRM) Underlie the Oncogenic Drive of SMARCA4 (BRG1) Mutation Boris G. Wilson,a Katherine C. Helming,a Xiaofeng Wang,a Youngha Kim,a Francisca Vazquez,c,d Zainab Jagani,f William C. Hahn,c,d,e Charles W. M. Robertsa,b,d,e Department of Pediatric Oncology, Dana-Farber Cancer Institute, Boston, Massachusetts, USAa; Division of Hematology/Oncology, Boston Children’s Hospital, Boston, Massachusetts, USAb; Department of Medical Oncology, Dana-Farber Cancer Institute, Boston, Massachusetts, USAc; Cancer Program, Broad Institute of Harvard and Massachusetts Institute of Technology, Boston, Massachusetts, USAd; Center for Cancer Genome Discovery, Dana-Farber Cancer Institute, Boston, Massachusetts, USAe; Novartis Institutes for BioMedical Research, Cambridge, Massachusetts, USAf Collectively, genes encoding subunits of the SWI/SNF (BAF) chromatin remodeling complex are mutated in 20% of all human Downloaded from cancers, with the SMARCA4 (BRG1) subunit being one of the most frequently mutated.