Three Membered Rings As Probes in the Activity Based Protein Profiling
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Cancer Drug Pharmacology Table
CANCER DRUG PHARMACOLOGY TABLE Cytotoxic Chemotherapy Drugs are classified according to the BC Cancer Drug Manual Monographs, unless otherwise specified (see asterisks). Subclassifications are in brackets where applicable. Alkylating Agents have reactive groups (usually alkyl) that attach to Antimetabolites are structural analogues of naturally occurring molecules DNA or RNA, leading to interruption in synthesis of DNA, RNA, or required for DNA and RNA synthesis. When substituted for the natural body proteins. substances, they disrupt DNA and RNA synthesis. bendamustine (nitrogen mustard) azacitidine (pyrimidine analogue) busulfan (alkyl sulfonate) capecitabine (pyrimidine analogue) carboplatin (platinum) cladribine (adenosine analogue) carmustine (nitrosurea) cytarabine (pyrimidine analogue) chlorambucil (nitrogen mustard) fludarabine (purine analogue) cisplatin (platinum) fluorouracil (pyrimidine analogue) cyclophosphamide (nitrogen mustard) gemcitabine (pyrimidine analogue) dacarbazine (triazine) mercaptopurine (purine analogue) estramustine (nitrogen mustard with 17-beta-estradiol) methotrexate (folate analogue) hydroxyurea pralatrexate (folate analogue) ifosfamide (nitrogen mustard) pemetrexed (folate analogue) lomustine (nitrosurea) pentostatin (purine analogue) mechlorethamine (nitrogen mustard) raltitrexed (folate analogue) melphalan (nitrogen mustard) thioguanine (purine analogue) oxaliplatin (platinum) trifluridine-tipiracil (pyrimidine analogue/thymidine phosphorylase procarbazine (triazine) inhibitor) -

IN-VITRO EVALUATION of the ANTICANCER ACTIVITY of Cu(II)AMINA(CYSTEINE)DITHIOCARBAMATE
Sys Rev Pharm 2020;11(9):43-51 A multifaceted review journal in the field of pharmacy IN-VITRO EVALUATION OF THE ANTICANCER ACTIVITY OF Cu(II)AMINA(CYSTEINE)DITHIOCARBAMATE Desy Kartina1, Abdul Wahid Wahab 2, Ahyar Ahmad3, Rizal Irfandi 4, Prihantono5, And Indah Raya6* 1Department of Chemistry, Faculty of Mathematics, and Natural Science, Hasanuddin University, Makassar 90245, Indonesia 2Department of Chemistry, Faculty of Mathematics, and Natural Science, Hasanuddin University, Makassar 90245, Indonesia 3Department of Chemistry, Faculty of Mathematics, and Natural Science, Hasanuddin University, Makassar 90245, Indonesia 4Department of Biology Education, Faculty of Teacher Training and Education, Puangrimaggalatung University Sengkang, 90915, Indonesia. 5Departement of Surgery, Faculty of Medicinal, Hasanuddin University, Makassar Indonesia, 90245 6*Department of Chemistry, Faculty of Mathematics, and Natural Science, Hasanuddin University, Makassar 90245, Indonesia ABSTRACT The Complex of Cu(II)cysteinedithiocarbamate has been synthesized, it Keywords: Anticancer, Cu(ll)Amina(cysteine)dithiocarbamate. was prepared by the “in situ method” and characterized by using Correspondance: Ultraviolet-Visible (UV-Vis), Infra-Red (IR) spectroscopy, X-Ray Indah Raya Fluorescence (XRF) instruments. While melting point and conductivity Department of Chemistry, also measured. The presence of UV-Vis maximum spectrums of Faculty of Mathematics, and Natural Science, Cu(II)cysteinedithiocarbamate at 296 nm and 436 nm indicated that Hasanuddin University, electronic transition π → π * dan n → π * of CS2 and N=C=S myotis. The Makassar 90245, Indonesia presence of the wavelength in the region of 399-540 cm-1 of IR spectra Email id : [email protected], [email protected] is indicated that has been coordination occurred between Cu(II) with Sulphur (S), Nitrogen (N), and Oxygen(O) atoms respectively from cysteinedithiocarbamate ligands. -

Recent Advances in Microwave-Assisted Copper-Catalyzed Cross-Coupling Reactions
catalysts Review Recent Advances in Microwave-Assisted Copper-Catalyzed Cross-Coupling Reactions Younis Baqi Department of Chemistry, College of Science, Sultan Qaboos University, P.O. Box 36, Muscat 123, Oman; [email protected]; Tel.: +968-2414-1473 Abstract: Cross-coupling reactions furnishing carbon–carbon (C–C) and carbon–heteroatom (C–X) bond is one of the most challenging tasks in organic syntheses. The early developed reaction protocols by Ullmann, Ullman–Goldberg, Cadiot–Chodkiewicz, Castro–Stephens, and Corey–House, utilizing elemental copper or its salts as catalyst have, for decades, attracted and inspired scientists. However, these reactions were suffering from the range of functional groups tolerated as well as severely restricted by the harsh reaction conditions often required high temperatures (150–200 ◦C) for extended reaction time. Enormous efforts have been paid to develop and achieve more sustainable reaction conditions by applying the microwave irradiation. The use of controlled microwave heating dramatically reduces the time required and therefore resulting in increase in the yield as well as the efficiency of the reaction. This review is mainly focuses on the recent advances and applications of copper catalyzed cross-coupling generation of carbon–carbon and carbon–heteroatom bond under microwave technology. Keywords: cross-coupling reaction; Cu catalyst; microwave irradiation; methodology; synthesis 1. Introduction Carbon–carbon (C–C) and carbon–heteroatom (C–X) bond formations through cross- Citation: Baqi, Y. Recent Advances in coupling reactions represents as one of the most useful strategy in the synthetic organic Microwave-Assisted chemistry, hence many procedures and methodologies have been developed and published Copper-Catalyzed Cross-Coupling in the literature. -

Copper ( II ) Chloride
Copyright © Tarek Kakhia. All rights reserved. http://tarek.kakhia.org Copper Contents 1 Introduction 2 History o 2.1 Copper Age o 2.2 Bronze Age o 2.3 Antiquity and Middle Ages o 2.4 Modern period 3 Characteristics o 3.1 Color o 3.2 Group 11 of the periodic table o 3.3 Occurrence o 3.4 Mechanical properties o 3.5 Electrical properties o 3.6 Corrosion . 3.6.1 Pure water and air/oxygen . 3.6.2 In contact with other metals . 3.6.3 Sulfide media . 3.6.4 Ammonia media . 3.6.5 Chloride media o 3.7 Germicidal effect o 3.8 Isotopes 4 Production 5 Applications o 5.1 Piping o 5.2 Electrical applications o 5.3 Architecture / Industry o 5.4 Household products o 5.5 Coinage o 5.6 Biomedical applications o 5.7 Chemical applications o 5.8 Other 6 Alloys 7 Compounds 8 Biological role 9 Recycling 1 Copyright © Tarek Kakhia. All rights reserved. http://tarek.kakhia.org 1 Introduction : Copper is a chemical element with the symbol Cu ( Latin : cuprum ) and atomic number 29 . It is a ductile metal with very high thermal and electrical conductivity. Pure copper is rather soft and malleable and a freshly - exposed surface has a pinkish or peachy color. It is used as a thermal conductor, an electrical conductor, a building material, and a constituent of various metal alloys. Copper metal and alloys have been used for thousands of years. In the Roman era, copper was principally mined on Cyprus, hence the origin of the name of the metal as Cyprium, "metal of Cyprus", later shortened to Cuprum. -

The Effect of High-Dose Thiotepa, Alone Or in Combination with Other
Bone Marrow Transplantation (2007) 40, 891–896 & 2007 Nature Publishing Group All rights reserved 0268-3369/07 $30.00 www.nature.com/bmt ORIGINAL ARTICLE The effect of high-dose thiotepa, alone or in combination with other chemotherapeutic agents, on a murine B-cell leukemia model simulating autologous stem cell transplantation A Abdul-Hai, L Weiss, D Ergas, IB Resnick, S Slavin and MY Shapira Department of Bone Marrow Transplantation and Cancer Immunotherapy, Hadassah–Hebrew University Medical Center, Jerusalem, Israel The use of thiotepa (TH) is increasing, especially in stem Introduction cell transplantation, mainly due to its safety and blood– brain barrier penetration. We evaluated the use of TH in a Thiotepa (TH, triethylene thiophosphoramide), an ethylene murine model simulating autologous stem cell transplan- amide, developed by Lederle Laboratories in 1952, tation, with or without additional agents. Between 1 and possesses mechlorethamine-like alkylating activity and 11 days following inoculation of BALB/c mice with 105– has been used clinically for over 35 years.1 It is of particular 108 B-cell leukemia (BCL1) cells (simulating pre-trans- use in breast cancer, mainly as second-line treatment.2 plant leukemia loads), each group received an ‘induction- TH has been given intrathecally for carcinomatous like’ irradiation and/or cytotoxic regimen. Animals were meningitis, and intravesically for bladder carcinoma.3,4 either followed without treatment, or an adoptive transfer Mild-to-moderate activity was observed in several solid (AT) was performed to untreated BALB/c mice. Admi- tumors and hematological malignancies.1,5 There has nistered alone without AT, high-dose TH did not change been, however, a sustained interest in defining clinical roles the time to appearance of leukemia. -

Thiirane-Terminated Polysulfide Polymers Thiiran-Terminierte Polysulfidpolymere Polymères Polysulfures À Terminaison Thiirane
(19) & (11) EP 2 121 807 B1 (12) EUROPEAN PATENT SPECIFICATION (45) Date of publication and mention (51) Int Cl.: of the grant of the patent: C08G 75/00 (2006.01) 26.05.2010 Bulletin 2010/21 (86) International application number: (21) Application number: 08707985.1 PCT/EP2008/050540 (22) Date of filing: 18.01.2008 (87) International publication number: WO 2008/090086 (31.07.2008 Gazette 2008/31) (54) THIIRANE-TERMINATED POLYSULFIDE POLYMERS THIIRAN-TERMINIERTE POLYSULFIDPOLYMERE POLYMÈRES POLYSULFURES À TERMINAISON THIIRANE (84) Designated Contracting States: • KOTTNER, Nils AT BE BG CH CY CZ DE DK EE ES FI FR GB GR D-78628 Rottweil (DE) HR HU IE IS IT LI LT LU LV MC MT NL NO PL PT • ZEITLER, Michael RO SE SI SK TR D-53347 Alfter (DE) (30) Priority: 23.01.2007 EP 07101024 (74) Representative: Schalkwijk, Pieter Cornelis et al 02.02.2007 US 899273 P Akzo Nobel N.V. Intellectual Property Department (43) Date of publication of application: P.O. Box 9300 25.11.2009 Bulletin 2009/48 6800 SB Arnhem (NL) (73) Proprietor: Akzo Nobel N.V. (56) References cited: 6824 BM Arnhem (NL) WO-A-03/076487 WO-A1-2004/099283 JP-A- 2004 062 057 US-A- 5 173 549 (72) Inventors: • WITZEL, Silke D-42103 Wuppertal (DE) Note: Within nine months of the publication of the mention of the grant of the European patent in the European Patent Bulletin, any person may give notice to the European Patent Office of opposition to that patent, in accordance with the Implementing Regulations. -

Copper Monosulfide (Cus) Powder SAFTY DATA SHEET
Copper Monosulfide (CuS) Powder US Research Nanomaterials, Inc. www.us-nano.com SAFTY DATA SHEET Revised Date 2/21/2016 1. PRODUCT AND COMPANY IDENTIFICATION 1.1 Product identifiers Product name: Copper Monosulfide (CuS) Powder Product Number : US1126M Copper Monosulfide (CuS) CAS#: 1317-40-4 1.2 Relevant identified uses of the substance or mixture and uses advised against Identified uses : Research 1.3 Details of the supplier of the safety data sheet Company: US Research Nanomaterials, Inc. 3302 Twig Leaf Lane Houston, TX 77084 USA Telephone: +1 832-460-3661 Fax: +1 281-492-8628 1.4 Emergency telephone number Emergency Phone # : (832) 359-7887 2. HAZARDS IDENTIFICATION 2.1 Classification of the substance or mixture This substance or mixture is not hazardous and is not classified under GHS. May cause irritation of skin, eyes, and respiratory tract. For the full text of the H-Statements mentioned in this Section, see Section 16. 2.2 GHS Label elements, including precautionary statements Pictogram Signal word None Hazard statement(s) H319 Causes serious eye irritation. H335 May cause respiratory irritation. Precautionary statement(s) P261 Avoid breathing dust/ fume/ gas/ mist/ vapors/ spray. P264 Wash skin thoroughly after handling. P271 Use only outdoors or in a well-ventilated area. P280 Wear protective gloves/ eye protection/ face protection. P304 + P340 IF INHALED: Remove victim to fresh air and keep at rest in a position comfortable for breathing. P305 + P351 + P338 IF IN EYES: Rinse cautiously with water for several minutes. Remove contact lenses, if present and easy to do. Continue rinsing. P312 Call a POISON CENTER or doctor/ physician if you feel unwell. -

SUMMARY of PARTICULARLY HAZARDOUS SUBSTANCES (By
SUMMARY OF PARTICULARLY HAZARDOUS SUBSTANCES (by alpha) Key: SC -- Select Carcinogens RT -- Reproductive Toxins AT -- Acute Toxins SA -- Readily Absorbed Through the Skin DHS -- Chemicals of Interest Revised: 11/2012 ________________________________________________________ ___________ _ _ _ _ _ _ _ _ _ _ _ ||| | | | CHEMICAL NAME CAS # |SC|RT| AT | SA |DHS| ________________________________________________________ ___________ | _ | _ | _ | _ | __ | | | | | | | 2,4,5-T 000093-76-5 | | x | | x | | ABRIN 001393-62-0 | | | x | | | ACETALDEHYDE 000075-07-0 | x | | | | | ACETAMIDE 000060-35-5 | x | | | | | ACETOHYDROXAMIC ACID 000546-88-3 ||x| | x | | ACETONE CYANOHYDRIN, STABILIZED 000075-86-5 | | | x | | x | ACETYLAMINOFLUORENE,2- 000053-96-3 | x | | | | | ACID MIST, STRONG INORGANIC 000000-00-0 | x | | | | | ACROLEIN 000107-02-8 | | x | x | x | | ACRYLAMIDE 000079-06-1 | x | x | | x | | ACRYLONITRILE 000107-13-1 | x | x | x | x | | ACTINOMYCIN D 000050-76-0 ||x| | x | | ADIPONITRILE 000111-69-3 | | | x | | | ADRIAMYCIN 023214-92-8 | x | | | | | AFLATOXIN B1 001162-65-8 | x | | | | | AFLATOXIN M1 006795-23-9 | x | | | | | AFLATOXINS 001402-68-2 | x | | x | | | ALL-TRANS RETINOIC ACID 000302-79-4 | | x | | x | | ALPRAZOMAN 028981-97-7 | | x | | x | | ALUMINUM PHOSPHIDE 020859-73-8 | | | x | | x | AMANTADINE HYDROCHLORIDE 000665-66-7 | | x | | x | | AMINO-2,4-DIBROMOANTHRAQUINONE 000081-49-2 | x | | | | | AMINO-2-METHYLANTHRAQUINONE, 1- 000082-28-0 | x | | | | | AMINO-3,4-DIMETHYL-3h-IMIDAZO(4,5f)QUINOLINE,2- 077094-11-2 | x | | | | | AMINO-3,8-DIMETHYL-3H-IMIDAZO(4,5-f)QUINOXALINE, -
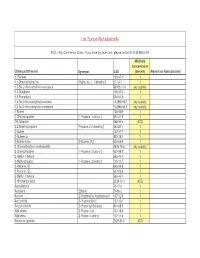
Homeland Security List
List Sorted Alphabetically ACG = Any Commercial Grade. If you have any questions, please contact EHS @ 898-5126. Minimum Concentration Chemical Of Interest Synonym CAS (percent) Amount on Hand (pounds) 1- Pentene 109-67-1 1 1,1-Dimethylhydrazine [Hydrazine, 1, 1-dimethyl-] 57-14-7 1 1,3-Bis(2-chloroethylthio)-n-propane 63905-10-2 any quantity 1,3-Butadiene 106-99-0 1 1,3-Pentadiene 504-60-9 1 1,4-Bis(2-chloroethylthio)-n-butane 142868-93-7 any quantity 1,5-Bis(2-chloroethylthio)-n-pentane 142868-94-8 any quantity 1-Butene 106-98-9 1 1-Chloropropylene [1-Propene, 1-chloro-] 590-21-6 1 1H-Tetrazole 288-94-8 ACG 2,2-Dimethylpropane [Propane, 2,2-dimethyl-] 463-82-1 1 2-Butene 107-01-7 1 2-Butene-cis 590-18-1 1 2-Butene-trans [2-Butene, (E)] 624-64-6 1 2-Chloroethylchloro-methylsulfide 2625-76-5 any quantity 2-Chloropropylene [1-Propene, 2-chloro-] 557-98-2 1 2-Methyl-1-butene 563-46-2 1 2-Methylpropene [1-Propene, 2-methyl-] 115-11-7 1 2-Pentene, (E)- 646-04-8 1 2-Pentene, (Z)- 627-20-3 1 3-Methyl-1-butene 563-45-1 1 5-Nitrobenzotriazol 2338-12-7 ACG Acetaldehyde 75-07-0 1 Acetylene [Ethyne] 74-86-2 1 Acrolein [2-Propenal] or Acrylaldehyde 107-02-8 1 Acrylonitrile [2-Propenenitrile] 107-13-1 1 Acrylyl chloride [2-Propenoyl chloride] 814-68-6 1 Allyl alcohol [2-Propen-1-ol] 107-18-6 1 Allylamine [2-Propen-1-amine] 107-11-9 1 Aluminum (powder) 7429-90-5 ACG Ammonia (anhydrous) 7664-41-7 1 Ammonia (conc. -

Synthesis and Structural Characterization of Semiconductors Based on Kesterites
Synthesis and Structural Characterization of Semiconductors based on Kesterites vorgelegt von Anna Ritscher, Bakk.rer.nat M.Sc. aus Villach, Österreich von der Fakultät II - Mathematik und Naturwissenschaften der Technischen Universität Berlin zur Erlangung des akademischen Grades Doktorin der Naturwissenschaften - Dr.rer.nat. - genehmigte Dissertation Promotionsausschuss: Vorsitzende: Prof. Dr. Regine von Klitzing Gutachter: Prof. Dr. Martin Lerch Gutachter: Prof. Dr. Manfred Mühlberg Tag der wissenschaftlichen Aussprache: 28.10.2016 Berlin, 2016 “There is a theory which states that if ever anyone discovers exactly what the Universe is for and why it is here, it will instantly disappear and be replaced by something even more bizarre and inexplicable. There is another theory mentioned, which states that this has already happened.” Douglas Adams, The Hitchhiker’s Guide to the Galaxy Abstract The here-presented thesis provides detailed investigations on the structural characteris- tics of the chalcogenide Cu2ZnSnS4 (CZTS). This semiconductor material is a promising candidate for absorber layers in solar cells due to its desirable properties for thin film photovoltaic applications. Yet, compared to current used chalcopyrite-based devices, effi- ciencies are significantly lower. This could be attributed to structural effects. Fundamental understanding of the structural characteristics of potential thin film absorber compounds is crucial for proper materials design in the field of solar technology. Therefore, intensive research is necessary to obtain knowledge on so far unexplored structural features or to certify and extend current literature data. The main objective of the here-presented work was to deepen the understanding of the quaternary sulfide 2Cu ZnSnS4. Primary, this implied the full characterization of structural properties of the compound using various diffraction techniques. -

Functionalisation of Colloidal Transition Metal Sulphides Nanocrystals: a Fascinating and Challenging Playground for the Chemist
crystals Review Functionalisation of Colloidal Transition Metal Sulphides Nanocrystals: A Fascinating and Challenging Playground for the Chemist Silvia Gross 1,2,*, Andrea Vittadini 1,2 and Nicola Dengo 1,2 1 Istituto di Chimica della Materia Condensata e di Tecnologie per l’Energia, ICMATE-CNR, via Marzolo 1, I-35131 Padova, Italy; [email protected] (A.V.); [email protected] (N.D.) 2 Dipartimento di Scienze Chimiche, Università degli Studi di Padova and INSTM, UdR Padova, via Marzolo 1, I-35131 Padova, Italy * Correspondence: [email protected]; Tel.: +39-049-827-5736 Academic Editor: Roberto Comparelli Received: 27 December 2016; Accepted: 24 March 2017; Published: 14 April 2017 Abstract: Metal sulphides, and in particular transition metal sulphide colloids, are a broad, versatile and exciting class of inorganic compounds which deserve growing interest and attention ascribable to the functional properties that many of them display. With respect to their oxide homologues, however, they are characterised by noticeably different chemical, structural and hence functional features. Their potential applications span several fields, and in many of the foreseen applications (e.g., in bioimaging and related fields), the achievement of stable colloidal suspensions of metal sulphides is highly desirable or either an unavoidable requirement to be met. To this aim, robust functionalisation strategies should be devised, which however are, with respect to metal or metal oxides colloids, much more challenging. This has to be ascribed, inter alia, also to the still limited knowledge of the sulphides surface chemistry, particularly when comparing it to the better established, though multifaceted, oxide surface chemistry. -
![Synthetic Approaches to Heterocyclic Bicyclo[2.1.0]Pentanes](https://docslib.b-cdn.net/cover/7157/synthetic-approaches-to-heterocyclic-bicyclo-2-1-0-pentanes-2177157.webp)
Synthetic Approaches to Heterocyclic Bicyclo[2.1.0]Pentanes
SYNTHETIC APPROACHES TO HETEROCYCLIC BICYCLO[2.1.0]PENTANES Rabah N. Alsulami A THESIS Submitted to the Graduate College of Bowling Green State University in partial fulfillment of The requirements for the degree of MASTER OF SCIENCE August 2011 Committee: Thomas H. Kinstle (Advisor) Marshall Wilson Alexander N. Tarnovsky ABSTRACT Thomas H. Kinstle, Advisor Bicyclic systems such as bicyclo[2.1.0]pentanes and 5-oxabicyclo[2.1.0]pentanes are known to display a variety of unique chemical properties associated with their high strain energy. To the best of our knowledge, there were no reports regarding synthesis and investigation of 5- azabicyclo[2.1.0]pentanes. Therefore, the initial goal of this research was synthesis of 5-azabicyclo[2.1.0]pentane and investigation of its chemical properties. The cycloaddition reaction of azides (58, 59, 61) to olefins (54, 55) with further elimination of nitrogen was chosen as a synthetic method in order to obtain the compounds of interest. Starting olefins (3,3-dimethyl-1-cyclobutene-1-carboxylic acid (54) and methyl 3,3-dimethyl-1-cyclobutene-1-carboxylate (55) and azides phenyl azide (58), p- toluenesulfonyl azide (59), and picryl azide (61) were successfully synthesized and characterized by NMR spectroscopy and GCMS spectrometry. The addition reaction between azides and olefins was performed under various conditions, such as different solvents and temperature; however, according to NMR spectroscopy and GCMS spectrometry, olefins (54, 55) do not undergo cycloaddition reaction with azides (58, 59, 61). In order to investigate that behavior, cycloaddition reactions of more reactive olefins (66, 68) with azides (58, 59, 61) were performed under a variety of conditions.