University Rawdah Q
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Six New Induced Sesquiterpenes from the Cultures of Ascomycete Daldinia Concentrica Xiang-Dong Qin, Hong-Jun Shao, Ze-Jun Dong, Ji-Kai Liu
J. Antibiot. 61(9): 556–562, 2008 THE JOURNAL OF ORIGINAL ARTICLE ANTIBIOTICS Six New Induced Sesquiterpenes from the Cultures of Ascomycete Daldinia concentrica Xiang-Dong Qin, Hong-Jun Shao, Ze-Jun Dong, Ji-Kai Liu Received: April 23, 2008 / Accepted: August 19, 2008 © Japan Antibiotics Research Association Abstract Six new sesquiterpenes having the botryane production titers of desired compounds. This approach was carbon skeleton (1ϳ6), together with known compounds successfully used as a valuable tool to exploit natural (7ϳ10) were induced and isolated from the ascomycete products diversity in the past, for example, actinomycetes Daldinia concentrica (strain S 0318). Structures (Streptomyces sp.) and fungi (Aspergillus sp., elucidation was accomplished by NMR spectroscopic and Sphaeropsidales sp.) produce additional compounds after X-ray crystallographic studies. variation of the culture conditions [3ϳ5]. Previous investigation reported the isolation of induced Keywords Sesquiterpenes, Daldinia concentrica, daldinins A, B, and C with a new skeleton and four known ascomycete, induction, botryane compounds from the cultures of ascomycete Daldinia concentrica [6]. Following the OSMAC approach, the strain S 0318 (Daldinia concentrica) has been cultivated Introduction and further investigated. In the culture broth extract from Erlenmeyer flasks (color, transparent; size, 500 ml; media, Fungi or bacteria that produce secondary metabolites often 300 ml) instead of reagent bottles under same cultivation have the potential to produce various compounds from a conditions we detected additional metabolites by TLC and single strain. Variation of cultivation parameters to induce HPLC. Careful investigation on the culture of D. the production of formerly unknown compounds is one of concentrica strain (S 0318) led to the isolation of new the approach to increase the number of secondary compounds methyl-7a-acetoxydeacetylbotryoloate (1), 7a- metabolites from one single organism. -

Studies on the Chemistry of Paclitaxel
STUDIES ON THE CHEMISTRY OF PACLITAXEL Haiqing Yuan Dissertation submitted to the Faculty of the Virginia Polytechnic Institute and State University in the partial fulfillment of the requirement for the degree of Doctor of Philosophy in Chemistry Dr. David G. I. Kingston, Chair Dr. Michael Calter Dr. Neal Castagnoli, Jr. Dr. Richard Gandour Dr. Larry Taylor August 11, 1998 Blacksburg, Virginia Keywords: Paclitaxel, Taxol®, synthesis, analog, SAR Copyright 1998, Haiqing Yuan STUDIES ON THE CHEMISTRY OF PACLITAXEL HAIQING YUAN (ABSTRACT) Paclitaxel is a natural occurring diterpene alkaloid originally isolated from the bark of Taxus brevifolia. It is now one of the most important chemotherapeutic agents for clinical treatment of ovarian and breast cancers. Recent clinical trials have also shown paclitaxel’s potential for the treatment of non-small-cell lung cancer, head and neck cancer, and other types of cancers. While tremendous chemical research efforts have been made in the past years, which established the fundamental structure-activity relationships of the paclitaxel molecule, and provided analogs for biochemical studies to elucidate the precise mechanism of action and for the development of second-generation agents, many areas remain to be explored. In continuation of our efforts in the structure-activity relationships study of A- norpaclitaxel, five new analogs modified at the C-1 substituent and analogs with expanded B-ring or contracted C-ring have now been prepared. Preliminary biological studies indicated that the volume rather than functionality at the C-1 position plays a role in determining the anticancer activity by controlling the relative position of the tetracyclic ring system, which in turn controls the positions of the most critical functionalities such as the C-2 benzoyl, the C-4 acetate, and the C-13 side chain. -

Acetoxy Drug: Protein Transacetylase: a Novel Enzyme-Mediating Protein Acetylation by Polyphenolic Peracetates*
Pure Appl. Chem., Vol. 77, No. 1, pp. 245–250, 2005. DOI: 10.1351/pac200577010245 © 2005 IUPAC Acetoxy drug: protein transacetylase: A novel enzyme-mediating protein acetylation by polyphenolic peracetates* Hanumantharao G. Raj1,‡, Brajendra K. Singh2, Ekta Kohli1, B. S. Dwarkanath3, Subhash C. Jain2, Ramesh C. Rastogi2, Ajit Kumar1, J. S. Adhikari3, Arthur C. Watterson4, Carl E. Olsen5, and Virinder S. Parmar2 1Biochemistry Department, V. P. Chest Institute, University of Delhi, Delhi 110 007, India; 2Bioorganic Laboratory, Department of Chemistry, University of Delhi, Delhi 110 007, India; 3Institute of Nuclear Medicine and Allied Sciences, Lucknow Road, Delhi 110 054, India; 4INSET, Department of Chemistry, University of Massachusetts, Lowell, MA 01854, USA; 5Department of Chemistry, Royal Veterinary and Agricultural University, DK-1871 Frederiksburg C, Copenhagen, Denmark Abstract: The acetylation of proteins in biological systems is largely catalyzed by specific acetyl transferases utilizing acetyl CoA as the acetyl donor. The enzymatic acetylation of pro- teins independent of acetyl CoA was unknown until we discovered a unique membrane- bound enzyme in mammalian cells catalyzing the transfer of acetyl groups from polypheno- lic peracetates (PAs) to certain enzyme proteins, resulting in the modulation of their catalytic activities. Since for the enzyme, acetyl derivatives of several classes of polyphenols such as coumarins, flavones, chromones, and xanthones were found to be acetyl donors, the enzyme was termed as acetoxy drug: protein transacetylase (TAase). TAase was found to be ubiqui- tously present in tissues of several animal species and a variety of animal cells. Liver micro- somal cytochrome P-450 (CYP), NADPH-cytochrome c reductase and cytosolic glutathione S-transferase (GST) were found to be the targets for TAase-catalyzed acetylation by the model acetoxy drug 7,8-diacetoxy-4-methylcoumarin (DAMC). -

"Point" of the Twist Danones.'4, 15 the Very Large Amplitudes
OPTICAL ROTATORY DISPERSION AND THE TWIST CONFORMATION OF CYCLOHEXANONES* BY CARL DJERASSI AND W. KLYNE CHEMISTRY DEPARTMENT, STANFORD UNIVERSITY, STANFORD, CALIFORNIA AND WESTFIELD COLLEGE. UNIVERSITY OF LONDON, LONDON, N.W. 3, ENGLAND Communicated April 25, 1962 The Octant Rule1 provides a theoretical basis for the study of optical rotatory dispersion curves of ketones. The application of this rule to extensive collections of data for cyclohexanone types from our two laboratories has been described else- where.2 3 Among the hundreds of curves recorded, a few stand out by reason of their unusually large amplitudes (positive or negative), which cannot be rationlized even in a roughly quantitative fashion by the usual octant treatment as applied to an all-chair conformation. Recently,4-8 there has been much interest in the existence of boat and modified boat conformations of cylohexane rings. Several authors9-11 have discussed forms intermediate between chairs and boats but the discussion has often been in rather general terms. However, Johnson et al.11 have recently provided thermodynamic evidence for the existence of an intermediate "twist" form in a lactone derived from trans-decalin. It is now suggested that this twist conformation provides a possible explanation of some of the grossly abnormal amplitudes found in decalone derivatives; the concept has also been used in dealing with some monocylic compounds.12 The twist conformation consists of two sets of contiguous coplanar C-atoms, 2, 1, 2' and 3, 4, 3'; C-1 and C4 are on the line of intersection of the two planes and may be called the "points" of the twist form. -
![[Beta]-Keto Sulfoxides Leo Arthur Ochrymowycz Iowa State University](https://docslib.b-cdn.net/cover/9355/beta-keto-sulfoxides-leo-arthur-ochrymowycz-iowa-state-university-2519355.webp)
[Beta]-Keto Sulfoxides Leo Arthur Ochrymowycz Iowa State University
Iowa State University Capstones, Theses and Retrospective Theses and Dissertations Dissertations 1969 Chemistry of [beta]-keto sulfoxides Leo Arthur Ochrymowycz Iowa State University Follow this and additional works at: https://lib.dr.iastate.edu/rtd Part of the Organic Chemistry Commons Recommended Citation Ochrymowycz, Leo Arthur, "Chemistry of [beta]-keto sulfoxides " (1969). Retrospective Theses and Dissertations. 3766. https://lib.dr.iastate.edu/rtd/3766 This Dissertation is brought to you for free and open access by the Iowa State University Capstones, Theses and Dissertations at Iowa State University Digital Repository. It has been accepted for inclusion in Retrospective Theses and Dissertations by an authorized administrator of Iowa State University Digital Repository. For more information, please contact [email protected]. This dissertation has been microfihned exactly as received 70-7726 OCHRYMOWYCZ, Leo Arthur, 1943- CHEMISTRY OF p -KETO SULFOXIDES. Iowa State University, Ph.D., 1969 Chemistry, organic University Microfilms, Inc., Ann Arbor, Michigan CHEMISTRY OF jg-KETO SULFOXIDES by Leo Arthur Ochrymowycz A Dissertation Submitted to the Graduate Faculty in Partial Fulfillment of The Requirements for the Degree of DOCTOR OF PHILOSOPHY Major Subject ; Organic Chemistry Approved : Signature was redacted for privacy. f Major Work Signature was redacted for privacy. d of Maj Department Signature was redacted for privacy. Graduate College Iowa State University Of Science and Technology Ames, Iowa 1969 il TABLE OP CONTENTS Page -

Synthesis and Characterization of Silicate Ester Prodrugs and Poly(Ethylene Glycol)
Synthesis and Characterization of Silicate Ester Prodrugs and Poly(ethylene glycol)-b-poly(lactic-co-glycolic acid) Block Copolymers for Formulation into Prodrug-Loaded Nanoparticles A DISSERTATION SUBMITTED TO THE FACULTY OF THE GRADUATE SCHOOL OF THE UNIVERSITY OF MINNESOTA BY Adam Richard Wohl IN PARTIAL FULFILLMENT OF THE REQUIREMENTS FOR THE DEGREE OF Doctor of Philosophy Thomas R. Hoye, Advisor September 2012 © Adam Richard Wohl September 2012 Acknowledgements I have been blessed to have the support of my family, friends, and colleagues. Without your help and encouragement, my graduate career would not have been possible. First of all, I would like to thank my advisor Prof. Tom Hoye. Over the past five years, he has been a consistent and patient mentor that leads first and foremost by setting a world-class example. He has taught me so much about analyzing problems, having fun while working hard, and always paying attention to the details. He has constantly taught me during our experimental discussions, results analysis, and late night (or, as I believe Tom would say, “mid-evening”) grant/manuscript writing sessions. I have learned not only many, many chemical concepts and scientific strategies, but also other valuable skills – how to clearly present ideas and make a compelling presentation to the right audience. Thank you, Tom, for all of the time that you have invested in both my scientific career and life – you have my utmost appreciation and respect. I would also like to thank Prof. Chris Macosko for all of his time, help, and expertise that he has provided to this collaborative project. -
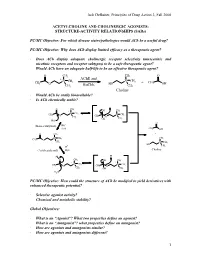
Jack Deruiter, Principles of Drug Action 2, Fall 2000 1 ACETYLCHOLINE and CHOLINERGIC AGONISTS: STRUCTURE-ACTIVITY RELATIONSHIPS
Jack DeRuiter, Principles of Drug Action 2, Fall 2000 ACETYLCHOLINE AND CHOLINERGIC AGONISTS: STRUCTURE-ACTIVITY RELATIONSHIPS (SARs) PC/MC Objective: For which disease states/pathologies would ACh be a useful drug? PC/MC Objective: Why does ACh display limited efficacy as a therapeutic agent? · Does ACh display adequate cholinergic receptor selectivity (muscarinic and nicotinic receptors and receptor subtypes) to be a safe therapeutic agent? · Would ACh have an adequate half-life to be an effective therapeutic agent? O CH3 CH3 O + AChE and + CH CH N CH3 N 3 + CH 3 O BuChE HO 3 OH CH3 CH3 Choline · Would ACh be orally bioavailable? · Is ACh chemically stable? O CH3 + OH CH3 + N CH3 CH3 N CH3 O CH3 CH3 O OH CH3 HO- O - (Base-catalyzed) HO H2O CH3 OH O CH3 + + N CH CH CH 3 3 3 O + CH3 N CH3 HO H+ CH3 (Acid-catalyzed) H2O Choline + H+ H O CH3 OH CH3 + + N CH3 N CH3 CH3 O CH3 CH O 3 OH CH3 H2O PC/MC Objective: How could the structure of ACh be modified to yield derivatives with enhanced therapeutic potential? · Selective agonist activity? · Chemical and metabolic stability? Global Objectives: · What is an “Agonist”? What two properties define an agonist? · What is an “Antagonist”? what properties define an antagonist? · How are agonists and antagonists similar? · How are agonists and antagonists different? 1 Jack DeRuiter, Principles of Drug Action 2, Fall 2000 MC Objective: Which ACh structural features appear to be required or at least contribute to cholinergic receptor binding. O CH3 + N CH3 CH3 O CH3 Ester Quaternary Ammonium MC Objective: -

Functionalization of Nanocellulouse Fibers for Use in Radical Reactions
The University of Maine DigitalCommons@UMaine Honors College Spring 2015 Functionalization of Nanocellulouse Fibers For Use In Radical Reactions Thomas McOscar University of Maine - Main, [email protected] Follow this and additional works at: https://digitalcommons.library.umaine.edu/honors Part of the Chemistry Commons Recommended Citation McOscar, Thomas, "Functionalization of Nanocellulouse Fibers For Use In Radical Reactions" (2015). Honors College. 237. https://digitalcommons.library.umaine.edu/honors/237 This Honors Thesis is brought to you for free and open access by DigitalCommons@UMaine. It has been accepted for inclusion in Honors College by an authorized administrator of DigitalCommons@UMaine. For more information, please contact [email protected]. FUNCTIONALIZATION OF NANOCELLULOSE FIBERS FOR USE IN RADICAL REACTIONS by Thomas McOscar A Thesis Submitted in Partial Fulfillment of the Requirements for a Degree with Honors (Chemistry) The Honors College University of Maine May 2015 Advisory Committee: William Gramlich, Assistant Professor of Chemistry, Advisor William DiSisto, Professor of Chemical Engineering Brian Frederick, Associate Professor of Chemistry Carl Tripp, Professor of Chemistry Kathleen Ellis, Adjunct Assistant Professor in Honors (English) Abstract Plastics and polymers comprise an expansive and growing portion of the materials in consumer products. The field of renewable and biodegradable polymers offers an appealing opportunity to continue developing plastics and materials along with an alternative to petroleum-based products that can have damaging environmental effects. Cellulose, the most abundant biological polymer on Earth, is an especially intriguing material for its diverse physical properties, its mass abundance, and the chemistry it can undergo, becoming a platform for other materials and reactions. -

Salvinorin A: a Potent Naturally Occurring Nonnitrogenous Opioid Selective Agonist
Salvinorin A: A potent naturally occurring nonnitrogenous opioid selective agonist Bryan L. Roth*†‡§¶, Karen Baner*, Richard Westkaemperሻ, Daniel Siebert**, Kenner C. Rice††, SeAnna Steinberg*, Paul Ernsberger*‡‡, and Richard B. Rothman§§ *National Institute of Mental Health Psychoactive Drug Screening Program, and Departments of †Biochemistry, ‡Psychiatry, §Neurosciences, and ‡‡Pharmacology and Nutrition, Case Western Reserve University Medical School, Cleveland, OH 44106; §§Clinical Psychopharmacology Section, Intramural Research Program, National Institute on Drug Abuse, National Institutes of Health, Baltimore, MD 21224; Department of Medicinal Chemistry, Medical College of Virginia, Richmond, VA 23298; **The Salvia divinorum Research and Information Center, Malibu, CA 90263; and ††Laboratory of Medicinal Chemistry, National Institute of Diabetes and Digestive and Kidney Diseases, National Institutes of Health, Bethesda, MD 20892 Edited by Erminio Costa, University of Illinois, Chicago, IL, and approved July 9, 2002 (received for review April 18, 2002) Salvia divinorum, whose main active ingredient is the neoclero- pharmacological properties of rodent and human molecular dane diterpene Salvinorin A, is a hallucinogenic plant in the mint targets are frequently distinct (7), and that tissue-based radio- family that has been used in traditional spiritual practices for its ligand binding assays frequently yield inaccurate estimates of psychoactive properties by the Mazatecs of Oaxaca, Mexico. More drug potency and selectivity. Accordingly, we reexamined the recently, S. divinorum extracts and Salvinorin A have become more molecular pharmacological profile of the novel diterpene Salvi- widely used in the U.S. as legal hallucinogens. We discovered that norin A at a large number of cloned human G protein-coupled Salvinorin A potently and selectively inhibited 3H-bremazocine receptors (GPCRs), channels, and transporters. -
![Golgi-Modifying Properties of Macfarlandin E and the Synthesis and Evaluation of Its 2,7-Dioxabicyclo[3.2.1]Octan-3-One Core](https://docslib.b-cdn.net/cover/3337/golgi-modifying-properties-of-macfarlandin-e-and-the-synthesis-and-evaluation-of-its-2-7-dioxabicyclo-3-2-1-octan-3-one-core-3853337.webp)
Golgi-Modifying Properties of Macfarlandin E and the Synthesis and Evaluation of Its 2,7-Dioxabicyclo[3.2.1]Octan-3-One Core
Golgi-modifying properties of macfarlandin E and the synthesis and evaluation of its 2,7-dioxabicyclo[3.2.1]octan-3-one core Martin J. Schnermanna, Christopher M. Beaudrya, Anastasia V. Egorovab, Roman S. Polishchukb,c, Christine Sütterlind,1, and Larry E. Overmana,1 aDepartment of Chemistry, 1102 Natural Sciences II, University of California, Irvine, CA 92697-2025; bTelethon Institute of Genetics and Medicine, Via P. Castellino 111, Naples, 8013, Italy; cTelethon Electron Microscopy Core Facility, Consorzio “Mario Negri Sud,” Santa Maria Imbaro (CH), 66030, Italy; and dDepartment of Developmental and Cell Biology, 2011 Biological Sciences III, University of California, Irvine, CA 92697-2300 Contributed by Larry E. Overman, February 4, 2010 (sent for review January 18, 2010) Golgi-modifying properties of the spongian diterpene macfar- landin E (MacE) and a synthetic analog, t-Bu-MacE, containing its 2,7-dioxabicyclo[3.2.1]octan-3-one moiety are reported. Natural product screening efforts identified MacE as inducing a novel morphological change in Golgi structure defined by ribbon frag- mentation with maintenance of the resulting Golgi fragments in the pericentriolar region. t-Bu-MacE, which possesses the substi- tuted 2,7-dioxabicyclo[3.2.1]octan-3-one but contains a tert-butyl group in place of the hydroazulene subunit of MacE, was prepared by chemical synthesis. Examination of the Golgi-modifying proper- Fig. 1. Known Golgi-modifying agents. ties of MacE, t-Bu-MacE, and several related structures revealed that the entire oxygen-rich bridged-bicyclic fragment is required for induction of this unique Golgi organization phenotype. Further specific localization of the Golgi is not required for general pro- characterization of MacE-induced Golgi modification showed that tein transport to the cell surface, but has recently been implicated protein secretion is inhibited, with no effect on the actin or micro- in directional protein transport in a polarized cell (18–20). -

Saul Winstein
NATIONAL ACADEMY OF SCIENCES S AUL WINSTEIN 1912—1969 A Biographical Memoir by W I L L I A M G. YO U N G A N D D O N A L D J . C RAM Any opinions expressed in this memoir are those of the author(s) and do not necessarily reflect the views of the National Academy of Sciences. Biographical Memoir COPYRIGHT 1973 NATIONAL ACADEMY OF SCIENCES WASHINGTON D.C. SAUL WINSTEIN October 8,1912-November 23,1969 BY WILLIAM G. YOUNG AND DONALD J. CRAM AUL WINSTEIN was born in Montreal, Canada, October 8, S 1912, the son of Louis and Anne Winstein. His death came suddenly at his home in West Los Angeles on November 23, 1969, at the age of fifty-seven, at the height of his career. He leaves his wife, Sylvia, whom he married on September 3, 1937; a son, Bruce, a graduate student at the California In- stitute of Technology; and a daughter, Carolee, a student at UCLA. Dr. Winstein came to the United States in 1923 and became a naturalized citizen in 1929. He graduated from Jefferson High School in Los Angeles in 1930, after which he received an A.B. degree in 1934 from the University of California at Los Angeles, followed by an M.A. degree in 1935 from the same institution. He received his Ph.D. degree in 1938 from the California Institute of Technology. After a postdoctoral fellowship at Cal-Tech, he spent 1939-1940 as a National Research Fellow at Harvard University, where he was associated with Professor Paul Bart- lett. -
![Crystal and Molecular Structure of a Benzo[A]Pyrene 7,8-Diol 9,10-Epoxide N2-Deoxyguanosine Adduct: Absolute Configuration and Conformation](https://docslib.b-cdn.net/cover/8396/crystal-and-molecular-structure-of-a-benzo-a-pyrene-7-8-diol-9-10-epoxide-n2-deoxyguanosine-adduct-absolute-configuration-and-conformation-4108396.webp)
Crystal and Molecular Structure of a Benzo[A]Pyrene 7,8-Diol 9,10-Epoxide N2-Deoxyguanosine Adduct: Absolute Configuration and Conformation
Crystal and molecular structure of a benzo[a]pyrene 7,8-diol 9,10-epoxide N2-deoxyguanosine adduct: Absolute configuration and conformation Isabella L. Karle*†, Haruhiko Yagi‡, Jane M. Sayer‡, and Donald M. Jerina‡ *Laboratory for the Structure of Matter, Naval Research Laboratory, Washington, DC 20375-5341; and ‡Laboratory of Bioorganic Chemistry, National Institute of Diabetes and Digestive and Kidney Diseases, Department of Health and Human Services, National Institutes of Health, Bethesda, MD 20892-0820 Contributed by Isabella L. Karle, December 18, 2003 Benzo[a]pyrene 7,8-diol 9,10-epoxide adducts in DNA are impli- to each other, the preferred conformation of the glycosidic bond cated in mutagenesis, and their formation from the diol epoxides between the sugar and the purine base, and the conformation of and subsequent incorrect replication by human DNA polymerases the partially saturated tetrahydro benzo-ring can potentially provide an attractive mechanism for the induction of cancer by this influence the fit of these adducts in DNA into the active site of highly carcinogenic hydrocarbon and its diol epoxide metabolites. DNA processing enzymes. Here, we describe the crystal structure of such an adduct at the Crystal structures for both the racemic BaP DE-1 (9) and exocyclic amino group of a purine nucleoside. The present adduct DE-2 (10) diastereomers have confirmed their relative stereo- derives from trans opening at C10 of the (؊)-(7S,8R)-diol (9R,10S)- chemistry as deduced by NMR (11, 12). Absolute configurations epoxide enantiomer by the exocyclic N2-amino group of deox- of their optically active precursor trans-7,8-dihydrodiols were yguanosine.