Title Mitochondrial Cytochrome B Phylogeny and Historical
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-
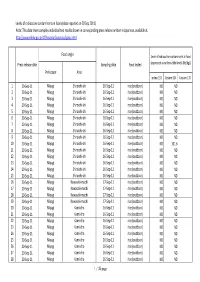
This Data Sheet Compiles Individual Test Results Shown in Corresponding
Levels of radioactive contaminants in foods (data reported on 20 Sep 2011) Note: This data sheet compiles individual test results shown in corresponding press release written in Japanese, available at http://www.mhlw.go.jp/stf/houdou/bukyoku/iyaku.html Food origin Level of radioactive contaminants in food Press release date Sampling date Food tested (expressed as radionuclide levels (Bq/kg)). Prefecture Area Iodine‐131 Cesium‐134 Cesium‐137 1 20‐Sep‐11 Miyagi Shiroishi‐shi 16‐Sep‐11 rice (outdoor) ND ND 2 20‐Sep‐11 Miyagi Shiroishi‐shi 16‐Sep‐11 rice (outdoor) ND ND 3 20‐Sep‐11 Miyagi Shiroishi‐shi 16‐Sep‐11 rice (outdoor) ND ND 4 20‐Sep‐11 Miyagi Shiroishi‐shi 16‐Sep‐11 rice (outdoor) ND ND 5 20‐Sep‐11 Miyagi Shiroishi‐shi 16‐Sep‐11 rice (outdoor) ND ND 6 20‐Sep‐11 Miyagi Shiroishi‐shi 16‐Sep‐11 rice (outdoor) ND ND 7 20‐Sep‐11 Miyagi Shiroishi‐shi 16‐Sep‐11 rice (outdoor) ND ND 8 20‐Sep‐11 Miyagi Shiroishi‐shi 16‐Sep‐11 rice (outdoor) ND ND 9 20‐Sep‐11 Miyagi Shiroishi‐shi 16‐Sep‐11 rice (outdoor) ND ND 10 20‐Sep‐11 Miyagi Shiroishi‐shi 16‐Sep‐11 rice (outdoor) ND 101.6 11 20‐Sep‐11 Miyagi Shiroishi‐shi 16‐Sep‐11 rice (outdoor) ND ND 12 20‐Sep‐11 Miyagi Shiroishi‐shi 16‐Sep‐11 rice (outdoor) ND ND 13 20‐Sep‐11 Miyagi Shiroishi‐shi 16‐Sep‐11 rice (outdoor) ND ND 14 20‐Sep‐11 Miyagi Shiroishi‐shi 16‐Sep‐11 rice (outdoor) ND ND 15 20‐Sep‐11 Miyagi Shiroishi‐shi 16‐Sep‐11 rice (outdoor) ND ND 16 20‐Sep‐11 Miyagi Kawasaki‐machi 17‐Sep‐11 rice (outdoor) ND ND 17 20‐Sep‐11 Miyagi Kawasaki‐machi 17‐Sep‐11 rice (outdoor) ND ND 18 20‐Sep‐11 Miyagi -

What Is Dewa Sanzan? the Spiritual Awe-Inspiring Mountains in the Tohoku Area, Embracing Peopleʼs Prayers… from the Heian Period, Mt.Gassan, Mt.Yudono and Mt
The ancient road of Dewa Rokujurigoegoe Kaido Visit the 1200 year old ancient route! Sea of Japan Yamagata Prefecture Tsuruoka City Rokujurigoe Kaido Nishikawa Town Asahi Tourism Bureau 60-ri Goe Kaido Tsuruoka City, Yamagata Prefecture The Ancient Road “Rokujuri-goe Kaido” Over 1200 years, this road has preserved traces of historical events “Rokujuri-goe Kaido,” an ancient road connecting the Shonai plain and the inland area is said to have opened about 1200 years ago. This road was the only road between Shonai and the inland area. It was a precipitous mountain road from Tsuruoka city to Yamagata city passing over Matsune, Juo-toge, Oami, Sainokami-toge, Tamugimata and Oguki-toge, then going through Shizu, Hondoji and Sagae. It is said to have existed already in ancient times, but it is not clear when this road was opened. The oldest theory says that this road was opened as a governmental road connecting the Dewa Kokufu government which was located in Fujishima town (now Tsuruoka city) and the county offices of the Mogami and Okitama areas. But there are many other theories as well. In the Muromachi and Edo periods, which were a time of prosperity for mountain worship, it became a lively road with pilgrims not only from the local area,but also from the Tohoku Part of a list of famous places in Shonai second district during the latter half of the Edo period. and Kanto areas heading to Mt. Yudono as “Oyama mairi” (mountain pilgrimage) custom was (Stored at the Native district museum of Tsuruoka city) booming. -

This Data Sheet Compiles Individual Test Results Shown in Corresponding
Levels of radioactive contaminants in foods reported on 16 - 22 June 2018 (Test results carried out since 1 April 2012) Note: This data sheet compiles individual test results shown in corresponding press release written in Japanese, available at http://www.mhlw.go.jp/stf/kinkyu/copy_of_copy_of_2r98520000016378.html Level of radioactive contaminants in Food origin Food Tested food (expressed as radionuclide levels (Bq/kg)). No Prefecture area Notes pre marketed/post marketed Food Category item name Notes Inspection instrument Sampling Date Results Obtained Date Cesium-134 Cesium-137 Cesium total 1 Iwate Otsuchi-cho - pre marketed livestock products Cattle meat - NaI 01-Jun-18 02-Jun-18 <15 <10 <25 2 Iwate Otsuchi-cho - pre marketed livestock products Cattle meat - NaI 01-Jun-18 02-Jun-18 <15 <10 <25 3 Iwate Ninohe-shi - pre marketed livestock products Cattle meat - NaI 01-Jun-18 02-Jun-18 <15 <10 <25 4 Iwate Ninohe-shi - pre marketed livestock products Cattle meat - NaI 01-Jun-18 02-Jun-18 <15 <10 <25 5 Iwate Ninohe-shi - pre marketed livestock products Cattle meat - NaI 01-Jun-18 02-Jun-18 <15 <10 <25 6 Iwate Ninohe-shi - pre marketed livestock products Cattle meat - NaI 01-Jun-18 02-Jun-18 <15 <10 <25 7 Iwate Yamada-machi - pre marketed livestock products Cattle meat - NaI 01-Jun-18 02-Jun-18 <15 <10 <25 8 Iwate Yamada-machi - pre marketed livestock products Cattle meat - NaI 01-Jun-18 02-Jun-18 <15 <10 <25 9 Iwate Yamada-machi - pre marketed livestock products Cattle meat - NaI 01-Jun-18 02-Jun-18 <15 <10 <25 10 Iwate Yamada-machi - -

Business Operation Plan for Fiscal Year Ending March 31, 2010
Business Operation Plan for Fiscal Year Ending March 31, 2010 February 27, 2009 Nippon Telegraph and Telephone East Corporation (“NTT East”) 1. Revenue and Expenses Plan (Unit: billions of yen) Item FY 3/10 FY 3/09 Change Operating Revenues 1,930 1,960 (30) Voice Transmission* (excluding IP-Related services) 820 910 (90) IP-Related* 660 575 85 Leased Circuits* (excluding IP- Related services) 160 171 (11) Operating Expenses 1,890 1,920 (30) Personnel* 114 115 (1) Non-Personnel* 1,245 1,280 (35) Depreciation, Amortization and Other* 531 525 6 Operating Income 40 40 0 Non-Operating Revenues 20 30 (10) Recurring Profits 60 70 (10) *Major items Note: Figures for FY 3/09 have changed since the results for the six months ended September 30, 2008 were announced. The figures announced for that period were 900 billion yen for voice transmission services (excluding IP-Related services) and 590 billion yen for IP-Related revenues. 1 2. Trends in Operating Income and Changes in Earnings Structure (Unit: Billions of yen) FY 3/07 FY 3/08 FY 3/09 Forecasts FY 3/10 Plan 59.9 Operating Income (14.9) 44.9 Operating Revenues 40.0 40.0 2,061.3 (4.9) ±0.0 (58.6) 2,002.7 (42.7) IP 1,960.0 (30.0) 1,930.0 359.4 +103.2 [17.4%] IP 462.6 Voice transmissions+IP +112.4 [23.1%] IP 1,518.5 IP (23.8) 575.0 +85.0 Voice transmissions +IP [29.3%] 660.0 1,494.6 [34.2%] (9.5) Voice transmissions +IP 1,485.0 (5.0) Voice transmissions +IP 1,480.0 Voice Transmissions 1,159.0 (127.0) Voice Transmissions 1,031.9 (121.9) Voice Transmissions 910.0 Voice Transmissions (90.0) 820.0 Other Other Other 542.8 Other 508.0 475.0 450.0 Note: Numbers in brackets indicate composition ratios of operating revenues 2 3. -

Note: This Data Sheet Compiles Individual Test Results Shown In
Levels of radioactive contaminants in foods reported on 23 - 29 June 2018 (Test results carried out since 1 April 2012) Note: This data sheet compiles individual test results shown in corresponding press release written in Japanese, available at http://www.mhlw.go.jp/stf/kinkyu/copy_of_copy_of_2r98520000016378.html Level of radioactive contaminants in Food origin Food Tested food (expressed as radionuclide levels (Bq/kg)). No Prefecture area Notes pre marketed/post marketed Food Category item name Notes Inspection instrument Sampling Date Results Obtained Date Cesium-134 Cesium-137 Cesium total 1 Yamagata Yamagata-shi - pre marketed livestock products Cattle meat NaI - 21-Jun-18 - - <25 2 Yamagata Sakata-shi - pre marketed livestock products Cattle meat NaI - 21-Jun-18 - - <25 3 Yamagata Sakata-shi - pre marketed livestock products Cattle meat NaI - 21-Jun-18 - - <25 4 Yamagata Sakata-shi - pre marketed livestock products Cattle meat NaI - 21-Jun-18 - - <25 5 Yamagata Shinjo-shi - pre marketed livestock products Cattle meat NaI - 21-Jun-18 - - <25 6 Yamagata Shinjo-shi - pre marketed livestock products Cattle meat NaI - 21-Jun-18 - - <25 7 Yamagata Sagae-shi - pre marketed livestock products Cattle meat NaI - 21-Jun-18 - - <25 8 Yamagata Sagae-shi - pre marketed livestock products Cattle meat NaI - 21-Jun-18 - - <25 9 Yamagata Tendo-shi - pre marketed livestock products Cattle meat NaI - 21-Jun-18 - - <25 10 Yamagata Tendo-shi - pre marketed livestock products Cattle meat NaI - 21-Jun-18 - - <25 11 Yamagata Tendo-shi - pre marketed livestock -

Digidepo 1164546 Po Nourinsu
The Report of the Study on the Protection of Cultural Landscapes Associated with Agriculture, Forestry and Fisheries Table of Contents Introduction 1 1. Background and Purpose of the Study 2 (1) Background 2 A. Academic Research on“Cultural Landscapes”in Japan 3 B. International Trend regarding“Cultural Landscapes”………………………………………… 4 (a)“Cultural Lanscapes”in the World Heritage Convention 4 Cultural Landscapes in the World Heritage Convention Rice Terraces of the Philippine Cordilleras Increasing Number of Inscriptions of“Cultural Landscapes”on the World Heritage List and Future Challenges (b) Protection of“Cultural Landscapes”in Other International Agreements 10 C. Rise in the Public Demand for the Protection of“Cultural Landscape”in Japan 10 (a) Reports of the National Council, etc. 11 Cultural Promotion Master Plan (Basic Plan) Reprort of the Cultural Properties Working Group of the Council for Cultural Affairs (b) Progress of Agricultural Land Conservation and Improvement Projects in Semi-Mountainous Regions 12 (c) Environmental Conservation in the Ancient Cities Preservation Law 13 (d) Promotion of“Cultural Landscapes”in Landscape Ordinances 14 D. Designation of“Cultural Landscapes”as Places of Scenic Beauty 17 (a)“Obasute (Tagoto no Tsuki)”…………………………………………………………… 17 (b)“Shiroyone no Semmaida”……………………………………………………………… 20 (2) Purpose and Methodology 21 2. History and Development of Study 21 (1) Definition of“Cultural Landscape” …………………………………………………………… 22 (2) 1st Phase Study and 2nd Phase Study …………………………………………………………… 22 (3) Categorization -

「Pre-Games Training Camp Facilities」 (Acceptable Only)
PyeongChang 2018 Olympic and Paralympic Games 「Pre-games Training Camp Facilities」 (Acceptable Only) TOHOKU / JAPAN No time difference Training Camps in KOREA PyeongChang 2018 TOHOKU ECONOMIC FEDERATION / JAPAN (February 2017 Edition) Convenient Railway & Airline in Tohoku Aomori → Seoul About 3 hours (By a direct flight) Aomori Prefecture Morioka PyeongChang → Aomori About 2018 Akita Prefecture Iwate Prefecture 50 minutes (By Shinkansen) Sendai → Seoul About 2.5 hours (By a direct flight) Sendai → Morioka About 40 minutes Miyagi Prefecture Niigata → Seoul Yamagata Prefecture (By Shinkansen) About 2.5 hours (By a direct flight) Niigata Prefecture Fukushima Prefecture Tokyo Tokyo → Sendai → Niigata About About 90 minutes 90 minutes Shinkansen (By Shinkansen) Tokyo → Seoul About 2.5 hours (By a direct flight) 2 / 19 Contents ・Alpine Skiing .................................. 4 ・Cross-country Skiing ...................... 7 ・Ski Jumping .................................... 9 ・Nordic Combined ...........................10 ・Freestyle Skiing ..............................11 ・Snowboard .....................................12 ・Speed Skating .................................14 ・Figure Skating .................................15 ・Short Track Speed Skating ..............16 ・Ice Hockey ......................................17 ・Curling ............................................18 ・Biathlon ..........................................19 「Pre-games Training Camp Facilities (Acceptable Only)」 TOHOKU / JAPAN Alpine Skiing You can see more detail information -

33. Zaozan (Zaosan))
(33. Zaozan (Zaosan)) 33. Zaozan (Zaosan) Continuously Monitored by JMA Latitude: 38°08'37" N, Longitude: 140°26'24" E, Elevation: 1,841 m (Kumanodake) (GSI Measuring Point) Overview of Zaozan (Zaosan). Taken from Zao Town on November 27, 2011 by the Japan Meteorological Agency. Summary Zaozan (Zaosan) is a basalt-andesite stratovolcano group, of which volcanic activity started with subaqueous eruption of the basaltic magma approximately 1 million to 700 thousand years ago. It was dormant for roughly 300,000 years, and then approximately 400,000 to 100,000 years ago many andesitic lava flows were discharged from multiple eruption centers, forming the peaks which make up the upper half of the volcanic edifice, such as Kumanodake (the highest peak) and Kattadake, forming the basic structure of the volcano. Approximately 30,000 years ago a caldera roughly 2 km in diameter was formed at the summit. At the same time, explosive activity involving basaltic andesite magma began, continuing intermittently even until today. Goshikidake is a post-caldera pyroclastic cone (or tuff cone) which formed within the caldera above. It has been active since approximately 2,000 years ago, and is the site of Okama crater lake (360 m in diameter, also known as Goshikinuma). Many records exist of eruptions at Okama within history. The eruptions which caused damage occurred at both inside and outside of Okama. Many of these eruptions were accompanied by lahars. Fumaroles are located in several areas, such as the new fumaroles to the northeast of Okama. The SiO2 content is between 51.3 and 64.1 wt %. -

A New Skull of an Early Diverging Rorqual (Balaenopteridae, Mysticeti, Cetacea) from the Late Miocene to Early Pliocene of Yamagata, Northeastern Japan
Palaeontologia Electronica palaeo-electronica.org A new skull of an early diverging rorqual (Balaenopteridae, Mysticeti, Cetacea) from the late Miocene to early Pliocene of Yamagata, northeastern Japan Yoshihiro Tanaka, Kazuo Nagasawa, and Yojiro Taketani ABSTRACT The family of rorquals and humpback whales, Balaenopteridae includes the larg- est living animal on Earth, the blue whale Balaenoptera musculus. Many new taxa have been named, but not many from the western Pacific, except Miobalaenoptera numataensis from Japan. Here we describe an early balaenopterid, cf. M. numataensis from a late Miocene to early Pliocene sediment in Yamagata Prefecture, northeastern Japan. The species has a straight and sharp lateral ridge of the fovea epitubaria at the ventral surface of the periotic, and a dorsoventrally thin pars cochlearis. The new spec- imen provides knowledge of supposed ontogenetic variation and periotic morphology in poorly known fossil balaenopterids. Yoshihiro Tanaka. Osaka Museum of Natural History, Nagai Park 1-23, Higashi-Sumiyoshi-ku, Osaka, 546- 0034, Japan. [email protected] Hokkaido University Museum, Kita 10, Nishi 8, Kita-ku, Sapporo, Hokkaido 060-0810 Japan, Numata Fossil Museum, 2-7-49, Minami 1, Numata town, Hokkaido 078-2225 Japan Kazuo Nagasawa. Yamagata Prefectural Touohgakkan Junior and Senior High School. 1-7-1 Chuo- Minami, Higashine City, Yamagata Prefecture, Japan 999-3730. [email protected] Yojiro Taketani. Aizuwakamatsu City, Fukushima Prefecture, Japan. [email protected] Keywords: rorquals; Balaenopteridae; Noguchi Formation; Furukuchi Formation; Miobalaenoptera numataensis; ontogenetic variation Submission: 25 May 2019. Acceptance: 20 February 2020. INTRODUCTION (Van Beneden, 1880; Strobel, 1881; Sacco, 1890; Bisconti, 2007a, 2007b, 2010; Bosselaers and The family of rorquals and humpback whales, Post, 2010; Bisconti and Bosselaers, 2016) and Balaenopteridae includes the largest living animal the East Coast of the U.S. -

Soak in Romantic Taishō-Era Japan a Panoramic View of Civilization and 12 Retracing the Steps of Poet Mokichi 6 Nature, Blended Harmoniously
Hiyoriyama Cherry Blossom Festival [Sakata-shi, Shōnai Region] Promoting the Scenic Beauty of Yamagata Enjoying the Sights of Yamagata There is more to be enjoyed at the views than what can be captured in a photograph. Learn more about each sight to make the experience all the more breathtaking. Go, see, feel, and spread the word! The prefecture of Yamagata is full of strikingly beautiful nature and rustic townscapes. We have selected 33 particularly recommended spots that we think everyone should have the Check which Sights to Visit Start by checking which sights were selected as Yamagata’ s 33 recommended sights. pleasure of seeing. In addition to its inherent scenic beauty, each spot can speak volumes There are two ways to do this. about its history or background. Also note that the sights mentioned here have parking lots 1) Visit the “Scenic Beauty of Yamagata” website from your computer or smartphone. nearby and are easily accessed by car, making it convenient to visit each and enjoy its surrounding areas. The Scenic Beauty of Yamagata URL:http://yamagata-keikanmonogatari.com “The Scenic Beauty of Yamagata” QR Code 2) View this pamphlet Basic information of each sight can be accessed by scanning the QR codes found by each Goal of this Project sight on pages 3-10 of this pamphlet. ● The goal of this project is to select and promote Yamagata’ s particularly beautiful scenic areas with the hopes of Visit the Sights and Find Information Signs increasing activity within the prefecture by revitalizing interest and encouraging interaction (restoring tourism) within the area. -

10 Years After the Great East Japan Earthquake Iwate
10 years after the Great East Japan Earthquake Iwate 200 Miyagi 100 Minister for Reconstruction Fukushima 0 Fukushima Daiichi(km) HIRASAWA Katsuei Nuclear Power Plant 22 February 2021 Kyoto Tokyo Great East Japan Earthquake (1) Scale (2) Damage (as of March 2020) - No. of human casualties: - Date: March 11, 2011 at 14:46 JST • Deceased 19,729 - Epicenter: 130 km off the Pacific Coast of • Missing 2,559 Tohoku Region - No. of damaged buildings: - Scale: Magnitude 9.0 • Completely destroyed 121,996 (The largest earthquake ever recorded in Japan) • Half destroyed 282,941 • Partially destroyed 748,461 Seismic intensity distribution (Seismic intensity from 4 to 7) Approx. 800km Tohoku Iwate Region Prefecture Miyagi Prefecture Fukushima Prefecture 2 Support from the World Int’l rescue teams operated in the devastated areas 163 countries and areas as well as 43 international organizations expressed their intention of assistance 24 countries and areas plus five international organizations dispatched rescue teams We have received relief supplies and donations from 128 countries, areas and organizations (Source: https://www.mofa.go.jp/j_info/visit/incidents/index.html ) 3 Ten years later: Tohoku Today and in the Future: https://www.reconstruction.go.jp/10year/en/index.html 4 Public housing for disaster-affected 100% 90% 80% 70% 60% Progress rate 50% of completion 40% 30% 20% 10% 0% End of End of End of End of End of End of End of End of End of FY2012 FY2013 FY2014 FY2015 FY2016 FY2017 FY2018 FY2019 FY2020 8 prefectures in disaster-affected areas Iwate Prefecture Miyagi Prefecture Fukushima Prefecture Otsuchi Town, Iwate Prefecture Ishinomaki City, Miyagi Prefecture 5 Projected opening of Reconstruction Roads & Reconstruction Support Roads (as of September 2020) Miyako-Morioka Connecting Road Sanriku Coastal Road National Rte. -
![Download Paper [PDF:2.0MB]](https://docslib.b-cdn.net/cover/3161/download-paper-pdf-2-0mb-4433161.webp)
Download Paper [PDF:2.0MB]
DPRIETI Discussion Paper Series 11-E-068 Do More Productive Firms Locate New Factories in More Productive Locations? An empirical analysis based on panel data from Japan's Census of Manufactures FUKAO Kyoji RIETI IKEUCHI Kenta NISTEP KIM YoungGak Senshu University KWON Hyeog Ug RIETI The Research Institute of Economy, Trade and Industry http://www.rieti.go.jp/en/ RIETI Discussion Paper Series 11-E-068 September 2011 Do More Productive Firms Locate New Factories in More Productive Locations? An empirical analysis based on panel data from Japan’s Census of Manufactures FUKAO Kyoji (Hitotsubashi University, RIETI) IKEUCHI Kenta (NISTEP) KIM YoungGak (Senshu University) KWON Hyeog Ug (Nihon University, RIETI) Abstract Using a Melitz-style model of heterogeneous firms, Baldwin and Okubo (2006) recently presented a theoretical model in which self-sorting occurs and more productive factories choose to locate in more productive areas. The model suggests that firm-specific factors and regional factors affect each other through the endogeneity of location decisions. However, to date there have been few studies empirically testing this issue. Against this background, our aim is to examine the relationship between firms and location-specific factors in location decisions using factory-level panel data from Japan’s Census of Manufactures. We begin by estimating how much of the differences in factories’ TFP levels can be explained by both firm and location effects. The estimation results show that both effects have a significant impact on the productivity level of a factory, and that the firm effects are more important than the location effects. We also find a statistically significant negative correlation between firm effects and location effects, and investigate what causes this relationship.