Facilitated Loading of Reca Protein Is Essential to Recombination by Recbcd Enzyme*
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Supplemental Methods
Supplemental Methods: Sample Collection Duplicate surface samples were collected from the Amazon River plume aboard the R/V Knorr in June 2010 (4 52.71’N, 51 21.59’W) during a period of high river discharge. The collection site (Station 10, 4° 52.71’N, 51° 21.59’W; S = 21.0; T = 29.6°C), located ~ 500 Km to the north of the Amazon River mouth, was characterized by the presence of coastal diatoms in the top 8 m of the water column. Sampling was conducted between 0700 and 0900 local time by gently impeller pumping (modified Rule 1800 submersible sump pump) surface water through 10 m of tygon tubing (3 cm) to the ship's deck where it then flowed through a 156 µm mesh into 20 L carboys. In the lab, cells were partitioned into two size fractions by sequential filtration (using a Masterflex peristaltic pump) of the pre-filtered seawater through a 2.0 µm pore-size, 142 mm diameter polycarbonate (PCTE) membrane filter (Sterlitech Corporation, Kent, CWA) and a 0.22 µm pore-size, 142 mm diameter Supor membrane filter (Pall, Port Washington, NY). Metagenomic and non-selective metatranscriptomic analyses were conducted on both pore-size filters; poly(A)-selected (eukaryote-dominated) metatranscriptomic analyses were conducted only on the larger pore-size filter (2.0 µm pore-size). All filters were immediately submerged in RNAlater (Applied Biosystems, Austin, TX) in sterile 50 mL conical tubes, incubated at room temperature overnight and then stored at -80oC until extraction. Filtration and stabilization of each sample was completed within 30 min of water collection. -

B Number Gene Name Mrna Intensity Mrna
sample) total list predicted B number Gene name assignment mRNA present mRNA intensity Gene description Protein detected - Membrane protein membrane sample detected (total list) Proteins detected - Functional category # of tryptic peptides # of tryptic peptides # of tryptic peptides detected (membrane b0002 thrA 13624 P 39 P 18 P(m) 2 aspartokinase I, homoserine dehydrogenase I Metabolism of small molecules b0003 thrB 6781 P 9 P 3 0 homoserine kinase Metabolism of small molecules b0004 thrC 15039 P 18 P 10 0 threonine synthase Metabolism of small molecules b0008 talB 20561 P 20 P 13 0 transaldolase B Metabolism of small molecules chaperone Hsp70; DNA biosynthesis; autoregulated heat shock b0014 dnaK 13283 P 32 P 23 0 proteins Cell processes b0015 dnaJ 4492 P 13 P 4 P(m) 1 chaperone with DnaK; heat shock protein Cell processes b0029 lytB 1331 P 16 P 2 0 control of stringent response; involved in penicillin tolerance Global functions b0032 carA 9312 P 14 P 8 0 carbamoyl-phosphate synthetase, glutamine (small) subunit Metabolism of small molecules b0033 carB 7656 P 48 P 17 0 carbamoyl-phosphate synthase large subunit Metabolism of small molecules b0048 folA 1588 P 7 P 1 0 dihydrofolate reductase type I; trimethoprim resistance Metabolism of small molecules peptidyl-prolyl cis-trans isomerase (PPIase), involved in maturation of b0053 surA 3825 P 19 P 4 P(m) 1 GenProt outer membrane proteins (1st module) Cell processes b0054 imp 2737 P 42 P 5 P(m) 5 GenProt organic solvent tolerance Cell processes b0071 leuD 4770 P 10 P 9 0 isopropylmalate -

Recd: the Gene for an Essential Third Subunit of Exonuclease V
Proc. Nati. Acad. Sci. USA Vol. 83, pp. 5558-5562, August 1986 Genetics recD: The gene for an essential third subunit of exonuclease V [genetic recombination/ceil viability/nudease-negative, recombination.-positive (recB*) phenotype/RecBCD enzyme] SUSAN K. AMUNDSEN*, ANDREW F. TAYLOR, ABDUL M. CHAUDHURYt, AND GERALD R. SMITH§ Fred Hutchinson Cancer Research Center, 1124 Columbia Street, Seattle, WA 98104 Communicated by Hamilton 0. Smith, February 24, 1986 ABSTRACT Exonuclease V (EC 3.1.11.5) of Escherichia The subunit composition of ExoV has been uncertain. coli, an enzyme with multiple activities promoting genetic Genetic complementation analysis of ExoV null mutants recombination, has previously been shown to contain two (those lacking any detectable activity) revealed two genes, polypeptides, the products of the recB and recC genes. We recB and recC (12). These genes code for two polypeptides report here that the enzyme contains in addition a third with approximate molecular masses of 120 and 110 kDa, polypeptide (a) with a molecular mass of about 58 kDa. The a respectively, seen in purified preparations of ExoV (8, 13, poypeptide is not synthesized by a class ofmutants (previously 14). ExoV can be dissociated by high salt and separated by designated recB*) lacking the nuclease activity of exonuclease column chromatography into two inactive fractions, desig- V but retaining recombination proficiency. The gene, recD, nated a and /; mixing a and f3 restores nuclease and ATPase coding for the a polypeptide is located near recB in the order activity (15, 16). Unexpectedly, a activity is present in thyA-recC-ptr-recB-recD-agA on the E. -

Supplementary Information
Supplementary information (a) (b) Figure S1. Resistant (a) and sensitive (b) gene scores plotted against subsystems involved in cell regulation. The small circles represent the individual hits and the large circles represent the mean of each subsystem. Each individual score signifies the mean of 12 trials – three biological and four technical. The p-value was calculated as a two-tailed t-test and significance was determined using the Benjamini-Hochberg procedure; false discovery rate was selected to be 0.1. Plots constructed using Pathway Tools, Omics Dashboard. Figure S2. Connectivity map displaying the predicted functional associations between the silver-resistant gene hits; disconnected gene hits not shown. The thicknesses of the lines indicate the degree of confidence prediction for the given interaction, based on fusion, co-occurrence, experimental and co-expression data. Figure produced using STRING (version 10.5) and a medium confidence score (approximate probability) of 0.4. Figure S3. Connectivity map displaying the predicted functional associations between the silver-sensitive gene hits; disconnected gene hits not shown. The thicknesses of the lines indicate the degree of confidence prediction for the given interaction, based on fusion, co-occurrence, experimental and co-expression data. Figure produced using STRING (version 10.5) and a medium confidence score (approximate probability) of 0.4. Figure S4. Metabolic overview of the pathways in Escherichia coli. The pathways involved in silver-resistance are coloured according to respective normalized score. Each individual score represents the mean of 12 trials – three biological and four technical. Amino acid – upward pointing triangle, carbohydrate – square, proteins – diamond, purines – vertical ellipse, cofactor – downward pointing triangle, tRNA – tee, and other – circle. -

Degradation of DNA RNA Hybrids by Ribonuclease H and DNA
Proc. Nat. Acad. Sci. USA Vol. 69, No. 11, pp. 3360-3364, November 1972 Degradation of DNA RNA Hybrids by Ribonuclease H and DNA Polymerases of Cellular and Viral Origin (RNA tumor viruses/DNA synthesis) WALTER KELLER* AND ROBERT CROUCHt * Cold Spring Harbor Laboratory, Cold Spring Harbor, Long Island, New York 11724; and t Laboratory of Molecular Genetics, National Institute of Child Health and Human Development, National Institutes of Health, Bethesda, Maryland 20014 Communicated by Barbara McClintock, September 7, 1972 ABSTRACT Ribonuclease H from human KB cells, (dT) were prepared by incubation of poly(dA) or poly(rA) chick embryos, calf thymus, avian myeloblastosis virus, (both from Miles Laboratories) at a concentration of 0.2 and Rous associated virus specifically degrades the RNA of DNA RNA hybrids, producing mono- and oligoribo- mg/ml with 0.05 mg/ml of oligo(dT) (chain length: 12-18 nucleotides terminated in 5'-phosphates. The cellular nucleotides, Collaborative Research, Inc.) in 0.1 M NaCl- RNase H is an endonuclease, whereas the viral enzyme 1 mM Na-EDTA (pH 7.6), for 30 min at 250. appears to be an exonuclease. Viral DNA polymerase and RNase H copurify through all separation steps. Therefore, Enzymes. DNA-Dependent RNA polymerase from Esche- RNase H activity is an intrinsic part of the viral DNA richia coli was purified by the procedure of Berg et al. (5), and polymerase. DNA RNA hybrids are also degraded by nu- cleases associated with cellular DNA polymerases and by was a gift from Dr. Michael Cashel. Exonuclease III from exonuclease III. However, these nucleases differ from E. -

Exonuclease III Digestion-Random Primer Extension Procedure
3232'Med Genet 1995;32:32-35 Difference in constitutive heterochromatin behaviour between human amniocytes and lymphocytes detected by a sequential in situ J Med Genet: first published as 10.1136/jmg.32.1.32 on 1 January 1995. Downloaded from exonuclease III digestion-random primer extension procedure J L Fernandez, A Campos, C L6pez-Fernandez, J Gosalvez, V Goyanes Abstract chromatin banding for certain human Fixed chromosomes from human amniotic chromosomes in not easily obtained. (3) The fluid cells and peripheral blood lympho- banding pattern resulting from a specific pro- cytes were digested in situ with exo- cedure is quite consistent across all human nuclease III and the single stranded DNA cellular types. obtained was used as template for an in In the present paper we present a modified situ random primer extension. Under in situ random primer extension procedure on these conditions an R banding pattern, exonuclease III treated chromosomes to show more evident in lymphocytes than in that there are exceptions to items (2) and (3) amniocytes, was obtained. Nevertheless, mentioned above. In fact, in the model we have constitutive heterochromatin of chro- tested, R bands coexist with labelled con- mosomes 1, 16, Yq, and mainly the peri- stitutive heterochromatin in chromosomes 1, centromeric region of chromosome 9 was 9, 16, and Yq. Moreover, this is shown to be far more intensely labelled in amniocytes specific to human amniocytes, human lympho- than in lymphocytes. Fluorescence in situ cytes producing a different banding pattern. -

The Recombination Hot Spot X Is a Regulatory Element That Switches the Polarity of DNA Degradation by the Recbcd Enzyme
Downloaded from genesdev.cshlp.org on September 30, 2021 - Published by Cold Spring Harbor Laboratory Press The recombination hot spot X is a regulatory element that switches the polarity of DNA degradation by the RecBCD enzyme Daniel G. Anderson I and Stephen C. Kowalczykowski 1-3 1Genetics Graduate Group, 2Sections of Microbiology and of Molecular and Cellular Biology, University of California at Davis, Davis, California 95616-8665 USA Homologous recombination in Escherichia coli is stimulated at DNA sequences known as X sites. Stimulation requires the multifunctional RecBCD enzyme, which is both a helicase and a 3' ~ 5' exonuclease. Upon recognition of a properly oriented X site, the 3' -9 5' exonuclease activity is attenuated. Here we show that in addition to attenuation of the 3' ~ 5' exonuclease activity, recognition of X by the RecBCD enzyme also up-regulates a nuclease activity of the opposite polarity, resulting in an enzyme that now preferentially degrades 5' ~ 3'. These results demonstrate that X is a unique regulatory element that converts the antirecombinogenic form of the RecBCD enzyme into a recombinogenic form by causing two distinct enzymatic changes: attenuation of the 3' ~ 5' nuclease activity, and up-regulation of the 5' ~ 3' nuclease activity. The consequence of X recognition is the production of a recombination intermediate possessing a 3'-ssDNA overhang terminating at the X sequence. This processing of a dsDNA end to a 3'-ssDNA overhang parallels that which occurs during the initation of homologous recombination in other pathways in E. coli, and in other organisms such as the yeast Saccharomyces cerevisiae. [Key Words: Recombination; RecBCD; X; nuclease; helicase; regulation] Received December 4, 1996; revised version accepted January 30, 1997. -

Characteristics of Genome Evolution in Obligate Insect Symbionts, Including The
Characteristics of genome evolution in obligate insect symbionts, including the description of a recently identified obligate extracellular symbiont. Thesis Presented in Partial Fulfillment of the Requirements for the Degree Master of Science in the Graduate School of The Ohio State University By Laura Jean Kenyon, B.S. Graduate Program in Evolution, Ecology, and Organismal Biology The Ohio State University 2015 Thesis Committee: Norman Johnson Andy Michel Kelly Wrighton Zakee L. Sabree, Advisor Copyright by Laura Jean Kenyon 2015 Abstract Animal-bacterial symbioses have shaped the evolution of all eukaryotic organisms. All symbioses have in common a long-term association and therefore provide valuable insight into the evolution and diversification of both partners. Insect-bacterial mutualisms represent the most extreme natural partnerships known, showing evidence of coevolution and obligate interdependence between the partners. Early investigations of plant sap-feeding insects, in particular, revealed tissues of unknown function inhabited by bacteria and insects void of their symbionts revealed reduced host fitness compared to symbiotic insects. Due to the obligate nature of the relationships, endosymbiotic bacteria are uncultivable, but complete genome sequencing suggests bacterial mutualists metabolic capabilities and likely contribution to the mutualisms, typically nutrient provisioning. A well-supported pattern of bacterial genome evolution for obligate mutualists is extreme genome reduction, likely due to relaxed selection upon genes that are not required in the stable environment of the host, leading to an accumulation of deleterious mutations in these genes, and eventually to their complete loss, leaving only those genes that are required for the relatively-stable life-style and for maintenance of the symbiosis. -

A Domain of Recc Required for Assembly of the Regulatory Recd Subunit Into the Escherichia Coli Recbcd Holoenzyme
Copyright 2002 by the Genetics Society of America A Domain of RecC Required for Assembly of the Regulatory RecD Subunit Into the Escherichia coli RecBCD Holoenzyme Susan K. Amundsen, Andrew F. Taylor and Gerald R. Smith1 Fred Hutchinson Cancer Research Center, Seattle, Washington 98109 Manuscript received January 9, 2002 Accepted for publication February 20, 2002 ABSTRACT The heterotrimeric RecBCD enzyme of Escherichia coli is required for the major pathway of double- strand DNA break repair and genetic exchange. Assembled as a heterotrimer, the enzyme has potent nuclease and helicase activity. Analysis of recC nonsense and deletion mutations revealed that the C terminus of RecC is required for assembly of the RecD subunit into RecBCD holoenzyme but not for recombination proficiency; the phenotype of these mutations mimics that of recD deletion mutations. Partial proteolysis of purified RecC polypeptide yielded a C-terminal fragment that corresponds to the RecD-interaction domain. RecD is essential for nuclease activity, regulation by the recombination hotspot Chi, and high affinity for DNA ends. The RecC-RecD interface thus appears critical for the regulation of RecBCD enzyme via the assembly and, we propose, disassembly or conformational change of the RecD subunit. NA repair and recombination are highly regulated loaded with RecA protein (Anderson and Kowalczy- D processes that require the modulation of degrada- kowski 1997). The RecA·ssDNA filament then under- tive and recombinogenic activities of multiple enzymes goes pairing and strand exchange with a homologous that act on DNA. Such regulation can alter the distribu- chromosome (Kowalczykowski 2000). The RecD sub- tion of recombination around special sites on the chro- unit plays at least two roles in the RecBCD enzyme-Chi mosome. -
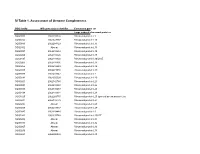
SI Table 1. Assessment of Genome Completeness
SI Table 1. Assessment of Genome Completeness COG family IMG gene object identifier Conserved gene set Large subunit ribosomal proteins COG0081 2062288324 Ribosomal protein L1 COG0244 2062347387 Ribosomal protein L10 COG0080 2062288323 Ribosomal protein L11 COG0102 Absent Ribosomal protein L13 COG0093 2062418832 Ribosomal protein L14 COG0200 2062418826 Ribosomal protein L15 COG0197 2062418838 Ribosomal protein L16/L10E COG0203 2062418836 Ribosomal protein L17 COG0256 2062418829 Ribosomal protein L18 COG0335 2062273558 Ribosomal protein L19 COG0090 2062418842 Ribosomal protein L2 COG0292 2062350539 Ribosomal protein L20 COG0261 2062142780 Ribosomal protein L21 COG0091 2062418840 Ribosomal protein L22 COG0089 2062138283 Ribosomal protein L23 COG0198 2062418834 Ribosomal protein L24 COG1825 2062269715 Ribosomal protein L25 (general stress protein Ctc) COG0211 2062142779 Ribosomal protein L27 COG0227 Absent Ribosomal protein L28 COG0255 2062418837 Ribosomal protein L29 COG0087 2062154483 Ribosomal protein L3 COG1841 2062335748 Ribosomal protein L30/L7E COG0254 Absent Ribosomal protein L31 COG0333 Absent Ribosomal protein L32 COG0267 Absent Ribosomal protein L33 COG0230 Absent Ribosomal protein L34 COG0291 2062350538 Ribosomal protein L35 COG0257 Absent Ribosomal protein L36 COG0088 2062138282 Ribosomal protein L4 COG0094 2062418833 Ribosomal protein L5 COG0097 2062418830 Ribosomal protein L6P/L9E COG0222 2062288326 Ribosomal protein L7/L12 COG0359 2062209880 Ribosomal protein L9 Small subunit ribosomal proteins COG0539 Absent Ribosomal protein -

Phosphatidylinositol-Specific Phospholipase C of Bacillus Cereus: Cloning, Sequencing, and Relationship to Other Phospholipases
JOURNAL OF BACTERIOLOGY, Nov. 1989, p. 6077-6083 Vol. 171, No. 11 0021-9193/89/116077-07$02.00/0 Copyright © 1989, American Society for Microbiology Phosphatidylinositol-Specific Phospholipase C of Bacillus cereus: Cloning, Sequencing, and Relationship to Other Phospholipases ANDREAS KUPPE,1 LOREENE M. EVANS,' DEBRA A. McMILLEN,2 AND 0. HAYES GRIFFITH'* Institute of Molecular Biology and Department of Chemistry,' and Biotechnology Laboratory,2 University of Oregon, Eugene, Oregon 97403 Received 11 May 1989/Accepted 10 August 1989 The phosphatidylinositol (PI)-specific phospholipase C (PLC) of Bacillus cereus was cloned into Escherichia coli by using monoclonal antibody probes raised against the purified protein. The enzyme is specific for hydrolysis of the membrane lipid PI and PI-glycan-containing membrane anchors, which are important structural components of one class of membrane proteins. The protein expressed in E. coli comigrated with B. cereus PI-PLC in sodium dodecyl sulfate-polyacrylamide gel electrophoresis, as detected by immunoblotting, and conferred PI-PLC activity on the host. This enzyme activity was inhibited by PI-PLC-specific monoclonal antibodies. The nucleotide sequence of the PI-PLC gene suggests that this secreted bacterial protein is synthesized as a larger precursor with a 31-amino-acid N-terminal extension to the mature enzyme of 298 amino acids. From analysis of coding and flanking sequences of the gene, we conclude that the PI-PLC gene does not reside next to the gene cluster of the other two secreted phospholipases C on the bacterial chromosome. The deduced amino acid sequence of the B. cereus PI-PLC contains a stretch of significant similarity to the glycosylphosphatidylinositol-specific PLC of Trypanosoma brucei. -

Exonuclease III Product Information Protocol 9PIM181
Certificate of Analysis Exonuclease III: Part No. Size (units) Part# 9PIM181 M181A 5,000 M181C 25,000 Revised 11/20 Exonuclease III 10X Reaction Buffer (E577A): The Exonuclease III 10X Reaction Buffer supplied with this enzyme has a composition of 660mM Tris-HCl (pH 8.0) and 6.6mM MgCl2. Enzyme Storage Buffer: Exonuclease III is supplied in 20mM Tris-HCl (pH 7.5), 1mM DTT, 100mM KCl and 50% glycerol. Source: E. coli cells expressing a recombinant clone. *AF9PIM181 1120M181* Unit Definition: The unit definition for Exonuclease III has been changed. One unit of Exonuclease III is defined as the amount of enzyme required to produce 1nmol of acid-soluble nucleotides from double-stranded DNA in 30 minutes at 37°C. See the AF9PIM181 1120M181 unit concentration on the Product Information Label. One unit as defined by these conditions is equivalent to one unit under the previous unit definition for this product. Storage Temperature: Store at –30°C to –10°C. Avoid multiple freeze-thaw cycles and exposure to frequent temperature changes. See the expiration date on the Product Information Label. Quality Control Assays Activity Assay 3´-Overhang Protection Assay: Twenty-five units of Exonuclease III are incubated with 1µg of Pst I cut plasmid at 37°C for Promega Corporation 4 minutes. The plasmid DNA shows <10% degradation when visualized by ethidium bromide-stained agarose gel electrophoresis. 2800 Woods Hollow Road Madison, WI 53711-5399 USA Contaminant Activity Telephone 608-274-4330 Endonuclease Assay: To test for endonuclease activity, 1µg of Type I supercoiled plasmid DNA is incubated with 50 units of Toll Free 800-356-9526 Fax 608-277-2516 Exonuclease III in 50mM Tris-HCl (pH 7.6), 10mM MgCl2, 1mM DTT for 1 hour at 37°C.