Flow Cytometry Protocols Flow Cytometry Protocols
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Supplementary Data for Publication
Electronic Supplementary Material (ESI) for Physical Chemistry Chemical Physics. This journal is © the Owner Societies 2016 Supplementary Data for Publication Synthesis of Eucalyptus/Tea Tree Oil Absorbed Biphasic Calcium phosphate-PVDF Polymer Nanocomposite Films: A Surface Active Antimicrobial System for Biomedical Application Biswajoy Bagchi1,δ, Somtirtha Banerjee1, Arpan Kool1, Pradip Thakur1,2, Suman Bhandary3, Nur Amin Hoque1 , Sukhen Das1+* 1Physics Department, Jadavpur University, Kolkata-700032, India. 2Department of Physics, Netaji Nagar College for Women, Kolkata-700092, India. 3Division of Molecular Medicine, Bose Institute, Kolkata-700054, India. +Present Address: Department of Physics, Indian Institute of Engineering Science and Technology, Shibpur, Howrah, West Bengal-711103, India. §Present Address: Fuel Cell and Battery Division, Central Glass and Ceramic Research Institute, Kolkata-700032, India. *Corresponding author’s email id: [email protected] Contact: +919433091337 Antimicrobial activity of EU and TTO treated films on E .coli and S. aureus by acridine orange/ethidium bromide (AO/EB) dual staining Live/dead cell characterization of EU/TTO film treated bacterial cultures was also done to visualize the viability under fluorescence microscope (). The treated culture suspensions after 12 and 24 hours of incubation were collected by centrifugation (5000 rpm, 20 mins). The cell pellets were resuspended in PBS. The staining solution was prepared by mixing equal parts of acridine orange (5mg/mL) and ethidium bromide (3mg/mL) in ethanol. 20μL of the staining solution is then mixed with 10μL of the resuspended solution and incubated for 15 minutes at 37°C. 10μL of this solution was then placed on a glass slide and covered with cover slip to observe under fluorescence microscope. -

Novel Acridine Orange Staining Protocol and Microscopy with UV Surface Excitation Allows for Rapid Histological Assessment of Canine Cutaneous Mast Cell Tumors
Novel Acridine Orange Staining Protocol and Microscopy with UV Surface Excitation Allows for Rapid Histological Assessment of Canine Cutaneous Mast Cell Tumors Croix Griffin1, Richard Levenson2, Farzad Feredouni2, Austin Todd2 1. School of Veterinary Medicine, University of California, Davis 2. Department of Pathology and Laboratory Medicine, University of California, Davis Compression Sponge Tissue Introduction ❖ XYZ Stage Stained for 1 minute UV window ❖ Mast cell tumors (MCTs) represent up to 27% of all malignant cutaneous tumors in with 0.03% Acridine dogs. Diagnosis is easily made with cytology, however the grade of the tumor LED: 1 Orange titrated to determines prognosis and required margins. UV excitation pH 0.53 with HCl ~280 nm ❖ Grade is not typically known until at least 48 hours after the surgery, as pre-surgical incisional biopsies are impractical due to additional cost and risk to the ❖ Rinsed in phosphate Objective patient. 5, 10, or 20X buffered saline for 1 ❖ Up to 30% of all mid to high grade mast cell tumors recur when incompletely minute at 50°C Tube Lens excised. Recurrence is correlated with progression to malignancy in 59% of patients.2 Figure 1. Example of MCT removal surgery. Figure 2. Blue arrow route does Figure 3. A streamlined, robust, Figure 4. MUSE Microscope. Please ❖ Low grade tumors are unlikely to recur regardless of margin cleanliness.3 When Single or multiple biopsies can be cut by hand not require formalin fixation or yet relatively gentle MC staining visit www.musepathology.com for removed with wide margins, the patient endures unnecessary short term morbidity from the tumor bed or tumor itself. -

ACRIDINE ORANGE STAIN - for in Vitro Use Only - Catalogue No
ACRIDINE ORANGE STAIN - For in vitro use only - Catalogue No. SA16 Our Acridine Orange Stain is used as a Quality Control fluorescent staining agent to detect the presence of bacteria in blood cultures and other bodily fluids. After checking for correct pH, colour, depth, Acridine orange is a fluorochrome dye that and sterility, the following organisms are used to can interchalate into nucleic acid. At a low pH determine the growth performance of the under UV light, bacterial and fungal nucleic acid completed medium. fluoresces orange whereas background mammalian nucleic acid fluoresces green. This Organism Expected Results rapid fluorescent staining procedure has been reported to be more sensitive than the Gram Escherichia coli Orange fluorescence staining procedure in the detection of ATCC 25922 microorganisms in blood cultures, cerebral spinal fluid and buffy coat preparations. Acridine orange stain can also aid in the detection of Storage and Shelf Life Acanthamoeba infections, infectious keratitis, Helicobacter pylori gastritis, and cell wall Our Acridine Orange Stain should be stored in deficient-bacteria such as Mycoplasma . the upright position at room temperature. Under these conditions this medium has a shelf life of 52 Formulation per Litre weeks from the date of manufacture. Acridine Orange ......................................... 100 mg Acetate Buffer .......................................... 1000 mL Ordering Information pH 4.0 ± 0.2 Cat# Description Format SA16-250 Acridine Orange Stain 250-mL Each Recommended Procedure 1. The prepared slide is fixed in methanol and air-dried. References 2. Flood the slide with Acridine Orange Stain. Allow the stain to sit on the slide for 2 1. Lauer BA, Reller LB, Mirrett S. -

Binding of Acridine Orange to DNA in Situ of Cells from Patients with Acute Leukemia1
(CANCER RESEARCH 49. .1692-3695. July 1. 1989] Binding of Acridine Orange to DNA in Situ of Cells from Patients with Acute Leukemia1 Alexander J. Walle2 and George Y. Wong Cornell Õ'nirersily Medical (allege /A. J. H'./and Memorial Sloan-Kettering Cancer Ccaler ¡G.Y. W.], New York, New York 10021 ABSTRACT cytes bound AO in a manner significantly different from that of human leukemic blood lymphoblasts [L3 variety of the Fluorescence flow cytometry was used to generate DNA histograms of French-American-British classification (6)]. We quantitatively at nilim- orange stained leukemic cell populations in G(I-G| phase of the described the interaction of AO with DNA of leukemic cell cell cycle. Complexes of the intercalating agent, acridine orange, with populations in G0-G| phase of the cell cycle by measuring the double-stranded DNA in situ, emit green fluorescence upon excitation with blue laser light. The histograms were evaluated by tirsi determining standard deviation of green fluorescence intensity pulses emit ted from AO-DNA complexes in single cells around the mean the standard deviation of the fluorescence intensity relative to the mean channel of fluorescence, i.e., the coefficient of variation, and then dividing channel of the fluorescence histograms. This represents the CV the coefficient of variation of a patient's sample by that of a control of the fluorescence intensity histograms (Fig. 1). To standardize sample (rCV). The mean rCV of cell populations of acute lymphoblastic and calculate the CVs of patient cell histograms, and subse leukemia (31 patients) differed significantly from that of nonlymphoblas- quently to analyze the quantitative differences between the tic leukemia (21 patients). -
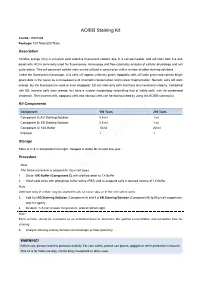
AO/EB Staining Kit
AO/EB Staining Kit Cat.No.: E607308 Package: 100 Tests/200 Tests Description Acridine orange (AO) is a nucleic acid selective fluorescent cationic dye. It is cell-permeable, and will stain both live and dead cells. AO is commonly used for fluorescence microscopy and flow cytometry analysis of cellular physiology and cell cycle status. This cell-permeant cellular stain can be utilized in conjunction with a number of other staining solutions. Under the fluorescent microscope: Live cells will appear uniformly green; Apoptotic cells will stain green and contain bright green dots in the nuclei as a consequence of chromatin condensation and nuclear fragmentation; Necrotic cells will stain orange, but the fluorescent is weak or even disappear. EB can stain only cells that have lost membrane integrity. Combined with EB, necrotic cells stain orange, but have a nuclear morphology resembling that of viable cells, with no condensed chromatin. Then normal cells, apoptotic cells and necrotic cells can be distinguished by using this AO/EB staining kit. Kit Components Component 100 Tests 200 Tests Component A: AO Staining Solution 0.5 ml 1 ml Component B: EB Staining Solution 0.5 ml 1 ml Component C: 10X Buffer 10 ml 20 ml Protocol 1 1 Storage Store at 2~8°C and protect from light. Reagent is stable for at least one year. Procedure Note: The follow procedure is adapted for most cell types. 1. Dilute 10X Buffer (Component C) with distilled water to 1X Buffer. 2. Wash cells twice with phosphate buffer saline (PBS) and re-suspend cells in desired volume of 1X Buffer. -

PGMD/Curcumin Nanoparticles for the Treatment of Breast Cancer Mankamna Kumari1, Nikita Sharma1, Romila Manchanda2, Nidhi Gupta3, Asad Syed4, Ali H
www.nature.com/scientificreports OPEN PGMD/curcumin nanoparticles for the treatment of breast cancer Mankamna Kumari1, Nikita Sharma1, Romila Manchanda2, Nidhi Gupta3, Asad Syed4, Ali H. Bahkali4 & Surendra Nimesh1* The present study aims at developing PGMD (poly-glycerol-malic acid-dodecanedioic acid)/curcumin nanoparticles based formulation for anticancer activity against breast cancer cells. The nanoparticles were prepared using both the variants of PGMD polymer (PGMD 7:3 and PGMD 6:4) with curcumin (i.e. CUR NP 7:3 and CUR NP 6:4). The size of CUR NP 7:3 and CUR NP 6:4 were found to be ~ 110 and 218 nm with a polydispersity index of 0.174 and 0.36, respectively. Further, the zeta potential of the particles was − 18.9 and − 17.5 mV for CUR NP 7:3 and CUR NP 6:4, respectively. The entrapment efciency of both the nanoparticles was in the range of 75–81%. In vitro anticancer activity and the scratch assay were conducted on breast cancer cell lines, MCF-7 and MDA-MB-231. The IC50 of the nanoformulations was observed to be 40.2 and 33.6 μM at 48 h for CUR NP 7:3 and CUR NP 6:4, respectively, in MCF-7 cell line; for MDA-MB-231 it was 43.4 and 30.5 μM. Acridine orange/EtBr and DAPI staining assays showed apoptotic features and nuclear anomalies in the treated cells. This was further confrmed by western blot analysis that showed overexpression of caspase 9 indicating curcumin role in apoptosis. With the changing environmental factors and lifestyle such as pollution, tobacco smoking, diet patterns, there has been a tremendous increase in the incidence of cancer. -

Cytotoxicity and Antiviral Activity of Palladium(II) and Platinum(II) Complexes with 2-(Diphenylphosphino)Benzaldehyde 1-Adamantoylhydrazone
Turkish Journal of Biology Turk J Biol (2016) 40: 661-669 http://journals.tubitak.gov.tr/biology/ © TÜBİTAK Research Article doi:10.3906/biy-1503-23 Cytotoxicity and antiviral activity of palladium(II) and platinum(II) complexes with 2-(diphenylphosphino)benzaldehyde 1-adamantoylhydrazone 1, 2 3 3 2 Vera SIMIĆ *, Stoimir KOLAREVIĆ , Ilija BRČESKI , Dejan JEREMIĆ , Branka VUKOVIĆ-GAČIĆ 1 National Control Laboratory, Medicines and Medical Devices Agency of Serbia, Belgrade, Serbia 2 Chair of Microbiology, Center for Genotoxicology and Ecogenotoxicology, Faculty of Biology, University of Belgrade, Belgrade, Serbia 3 Faculty of Chemistry, University of Belgrade, Belgrade, Serbia Received: 09.03.2015 Accepted/Published Online: 26.08.2015 Final Version: 18.05.2016 Abstract: Metal coordination compounds have an important role in the development of novel drugs. Using the resazurin microtitration assay we assessed the cytotoxicity and antiviral activity of the ligand 2-(diphenylphosphino)benzaldehyde 1-adamantoylhydrazone and its Pd(II) and Pt(II) complexes. Cytotoxicity was tested in A549 human lung adenocarcinoma epithelial cells. We observed that the ligand displayed a more pronounced cytotoxic activity than the platinum-based drug, carboplatin. Morphological evaluation of A549 cells treated with the ligand by acridine orange and ethidium bromide double staining revealed the presence of signs of apoptosis. Antiviral activity against poliovirus type 1 was assessed by examination of the cytopathic effect (CPE) in Hep-2 cells. Cells that were exposed to the 19 µM ligand before infection displayed a maximal significant reduction (by 24.42 ± 1.49%) of the CPE. This was likely due to the inhibition of virus receptors and prevention of viral adsorption. -

Developing Fluorogenic Reagents for Detecting and Enhancing Bloody Fingerprints
The author(s) shown below used Federal funds provided by the U.S. Department of Justice and prepared the following final report: Document Title: Developing Fluorogenic Reagents for Detecting and Enhancing Bloody Fingerprints Author: Robert M. Strongin, Dr.Martha Sibrian-Vazquez Document No.: 227841 Date Received: August 2009 Award Number: 2007-DN-BX-K171 This report has not been published by the U.S. Department of Justice. To provide better customer service, NCJRS has made this Federally- funded grant final report available electronically in addition to traditional paper copies. Opinions or points of view expressed are those of the author(s) and do not necessarily reflect the official position or policies of the U.S. Department of Justice. Developing Fluorogenic Reagents for Detecting and Enhancing Bloody Fingerprints Award 2007-DN-BX-K171 Authors Prof. Robert M. Strongin Dr.Martha Sibrian-Vazquez 1 This document is a research report submitted to the U.S. Department of Justice. This report has not been published by the Department. Opinions or points of view expressed are those of the author(s) and do not necessarily reflect the official position or policies of the U.S. Department of Justice. Abstract Fingerprints are the most common and useful physical evidence for the apprehension and conviction of crime perpetrators. Fluorogenic reagents for detecting and enhancing fingerprints in blood, however, have several associated challenges. For instance, they are generally unsuitable for dark and multi-colored substrates. Luminol and fluorescin and other chemilumigens and fluorigens can be used with dark and often multi-colored substrates, but are not compatible with fixatives and their oxidation products are not insoluble. -

Use of SYBR Green I for Rapid Epifluorescence Counts of Marine Viruses and Bacteria
AQUATIC MICROBIAL ECOLOGY Published February 13 Aquat Microb Ecol Use of SYBR Green I for rapid epifluorescence counts of marine viruses and bacteria Rachel T. Noble*,Jed A. Fuhrman University of Southern California, Department of Biological Sciences, AHF 107, University Park. Los Angeles, California 90089-0371, USA ABSTRACT: A new nucleic acid stain, SYBR Green I, can be used for the rapid and accurate determi- nation of viral and bacterial abundances in diverse marine samples. We tested this stain with formalin- preserved samples of coastal water and also from depth profiles (to 800 m) from sites 19 and 190 km off- shore, by filtering a few m1 onto 0.02 pm pore-size filters and staining for 15 min. Comparison of bacterial counts to those made with acridine orange (AO) and virus counts with those made by trans- mission electron microscopy (TEM) showed very strong correlations. Bacterial counts with A0 and SYBR Green 1 were indistinguishable and almost perfectly correlated (r2= 0.99). Virus counts ranged widely, from 0.03 to 15 X 10' virus ml-l. Virus counts by SYBR Green 1were on the average higher than those made by TEM, and a SYBR Green 1 versus TEM plot yielded a regression slope of 1.28. The cor- relation between the two was very high with an value of 0.98. The precision of the SYBR Green I method was the same as that for TEM, with coefficients of variation of 2.9%. SYBR Green I stained viruses and bacteria are intensely stained and easy to distinguish from other particles with both older and newer generation epifluorescence microscopes. -

Acridine Orange/ Propidium Iodide Stain
Acridine Orange/ Product Description Propidium Iodide Stain Appearance Orange-red liquid F23001 Cell permeability Membrane permeable (acridine orange) Membrane impermeant (propidium iodide) Excitation/emission 500⁄526 nm (acridine orange) 533⁄617 nm (propidium iodide) Storage Acridine Orange/Propidium Iodide Stain is a cell viability dye that 4 °C in the dark causes viable nucleated cells to fluoresce green and nonviable nucleated cells to fluoresce red. Acridine Orange/Propidium Iodide Stain can be used to assess cell viability with the automated fluorescence cell counters of the LUNA™ family. Acridine orange permeates viable cells and binds to nucleic acids. Binding to dsDNA causes acridine orange to fluoresce green and binding to ssDNA or RNA causes it to fluoresce red. Propidium iodide binds to nucleic acids. Not being able to permeate intact cell membranes, propidium iodide is taken up by nonviable cells and cells with compromised membranes. Once bound to nucleic acids, its fluorescence increases 20-30 fold and causes the cell to fluoresce red. Due to Fö rster resonance energy transfer (FRET), the propidium iodide signal absorbs the acridine orange signal in nonviable cells, ensuring no double positive results. Directions for Use 1. Mix: 2 μL Acridine Orange/Propidium Iodide Stain 18 μL cell sample 2. Count the sample with a compatible LUNA™. Disclaimer This product is for research use only. Please consult the material safety data sheet for information regarding hazards and safe handling practices. Additional information is available on our website at www.logosbio.com. © Logos Biosystems 2018. All rights reserved. HEADQUARTERS FL 2 & 3 28 Simindaero 327beon-gil, Dongan-gu Anyang-si, Gyeonggi-do 14055 South Korea Tel: +82 (31) 478-4185 USA 7700 Little River Turnpike STE 207 Annandale, VA 22003 USA Tel: +1 (703) 622-4660, +1 (703) 942-8867 EUROPE 11B avenue de l’Harmonie 59650 Villeneuve d’Ascq France Tel: +33 (0)3 74 09 44 35 www.logosbio.com VL1801-01 . -

Project Information Manufacturer Information Contact Information
7881 114th Avenue North Evaluation of the Smiths Detection RespondeR™ RCI Largo, FL 33773 Raman Spectrometer ph (727) 549‐6067 www.nfstc.org Project Information Title: Evaluation of the Smiths Detection RespondeR™ RCI Raman Spectrometer Evaluation Type: Portable Raman Spectrometer Stakeholder: Smiths Detection Start Date: 05/10/2010 End Date: 09/01/10 Kit Model Number(s): 024-1001 Serial Number(s): 502601108E Cost: $30,000 standard Manufacturer Information Contact Information Manufacturer: Smiths Detection Mark Norman/ Randall Aramburu Phone Number: (203) 207-9700 Phone Number(s): (203) 207-9700 / (817) 562-4479 www.smithsdetection.com [email protected] [email protected] Evaluation Team Hillary Markert, Senior Forensic Specialist-Chemistry, 727.549.6067, ext. 179, [email protected] Joan Ring, Chemistry Technical Services Manager, 727.549.6067, ext. 154, [email protected] Nicole Campbell, Forensic Services Technical Associate, 727.549.6067, ext. 194, [email protected] Kirk Grates, Senior Forensic Specialist-Chemistry, 727.549.6067, ext. 179, [email protected] Evaluation Summary The Forensic Services Chemistry Section of the National Forensic Science Technology Center (NFSTC) performed an evaluation of the Smiths Detection RespondeR™ Raman spectrometer. This portable Raman spectrometer is currently used by law enforcement, border patrol officers, military personnel and other first responders to chemically characterize unknown solids, liquids, pastes, gels, and powders encountered in field environments. The evaluation included assessments of conformity, reproducibility, mixture sensitivity, specificity, portability, ruggedness, and ease of use, including sample preparation, library additions and training requirements. The objective of this product assessment was to provide data to agencies interested in incorporating portable Raman technology into their laboratory or field-testing protocols. -

A Novel Antioxidant Rich Compound 2-Hydoxy 4-Methylbenzaldehyde from Decalepis Arayalpathra Induces Apoptosis in Breast Cancer Cells
Biocatalysis and Agricultural Biotechnology 21 (2019) 101339 Contents lists available at ScienceDirect Biocatalysis and Agricultural Biotechnology journal homepage: http://www.elsevier.com/locate/bab A novel antioxidant rich compound 2-hydoxy 4-methylbenzaldehyde from Decalepis arayalpathra induces apoptosis in breast cancer cells Ramar Thangam a,*,1, Sivaraman Gokul b, Malairaj Sathuvan c, Veeraperumal Suresh c, Srinivasan Sivasubramanian a,** a King Institute of Preventive Medicine & Research, Chennai, Tamilnadu, India b School for Conservation of Natural Resources, Repository for Medicinal Resources, Trans-Disciplinary University (TDU-FRLHT), Bangalore, Karnataka, India c Centre for Advanced Studies in Botany, University of Madras, Chennai, Tamilnadu, India ARTICLE INFO ABSTRACT Keywords: Decalepis arayalpathra is an endangered and endemic medicinal plant, mainly used for its aromatic and ethno- Decalepis arayalpathra medical properties. In this study, we reported the biomedical potentials of 2-hydoxy 4-methylbenzaldehyde 2-Hydroxy 4-methylbenzaldehyde (2H4MB) from D. arayalpathra. The compound 2H4MB was isolated from fresh root barks of the plants using Cytotoxicity À steam-hydro distillation extraction method and quantifiedusing HPLC analysis, which yielded 6.5 � 0.8 μg g 1 of Antioxidant purifiedcompound. Further, the antioxidant and anticancer effects in vitro were studied in detail using MDA-MB- Cell cycle arrest ROS-Dependent apoptosis 231 cells by cell-based assays. 2H4MB was observed to display the features such as generation of reactive oxygen species (ROS), improved cytotoxicity, loss of mitochondrial membrane (Δψm) potential, and nuclear DNA frag mentations, cellular apoptosis and cell cycle arrest. Further, the bioavailability of 2H4MB may facilitate its use as a potential cancer therapeutic agent. 1. Introduction cases every year (Ferlay et al., 2015; Miller et al., 2016).