PDF (4 Chapter 2.Pdf)
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Dalton Transactions
Dalton Transactions View Article Online PAPER View Journal | View Issue The role of neutral Rh(PONOP)H, free NMe2H, boronium and ammonium salts in the dehydro- Cite this: Dalton Trans., 2019, 48, 14724 coupling of dimethylamine-borane using the 2 + cationic pincer [Rh(PONOP)(η -H2)] catalyst†‡ E. Anastasia K. Spearing-Ewyn,a Nicholas A. Beattie,b Annie L. Colebatch, a Antonio J. Martinez-Martinez, a Andrew Docker,a Timothy M. Boyd,a Gregg Baillie,b Rachel Reed,b Stuart A. Macgregor *b and Andrew S. Weller *a 1 F 3 The σ-amine-borane pincer complex [Rh(PONOP)(η -H3B·NMe3)][BAr 4][2, PONOP = κ -NC5H3-2,6- t 2 F (OP Bu2)2] is prepared by addition of H3B·NMe3 to the dihydrogen precursor [Rh(PONOP)(η -H2)][BAr 4], 1 F 1. In a similar way the related H3B·NMe2H complex [Rh(PONOP)(η -H3B·NMe2H)][BAr 4], 3, can be made in situ, but this undergoes dehydrocoupling to reform 1 and give the aminoborane dimer [H2BNMe2]2. Creative Commons Attribution 3.0 Unported Licence. NMR studies on this system reveal an intermediate neutral hydride forms, Rh(PONOP)H, 4, that has been prepared independently. 1 is a competent catalyst (2 mol%, ∼30 min) for the dehydrocoupling of H3B·Me2H. Kinetic, mechanistic and computational studies point to the role of NMe2H in both forming the neutral hydride, via deprotonation of a σ-amine-borane complex and formation of aminoborane, and Received 17th August 2019, closing the catalytic cycle by reprotonation of the hydride by the thus-formed dimethyl ammonium Accepted 3rd September 2019 + + [NMe2H2] . -

Elements of a Glovebox Glove Integrity Program
RESEARCH ARTICLE Elements of a Glovebox Glove Integrity Program Programmatic operations at the Los Alamos National Laboratory Plutonium Facility involve working with various amounts of plutonium and other highly toxic, alpha-emitting materials. The spread of radiological contamination on surfaces and airborne contamination and excursions of contaminants into the operator’s breathing zone are prevented through the use of a variety of gloveboxes. The glovebox gloves are the weakest part of this engineering control. As a matter of good business practice, a team of glovebox experts from Los Alamos National Laboratory has been assembled to proactively investigate processes and procedures that minimize unplanned openings in the glovebox gloves, i.e., breaches and failures. Working together, they have developed the key elements of an efficient Glovebox Glove Integrity Program (GGIP). In the following report, the consequences of a glove failure or breach are identified, the acceptable risk is clarified, and elements needed to implement an efficient GGIP are discussed. By Michael E. Cournoyer, INTRODUCTION Through an integrated approach, Julio M. Castro, controls have been developed and Michelle B. Lee, Plutonium requires a high degree of implemented that come from input Cindy M. Lawton, confinement and continuous control from glovebox workers, scientists, Young Ho Park, measures in nuclear research labora- health physicists, statisticians, and phy- Roy Lee, tories because of its extremely low sical therapists. Working together, they Stephen Schreiber permissible body burden.1 Methods have developed an efficient Glovebox and equipment must be designed Glove Integrity Program (GGIP). toward the ultimate accomplishment Recent accomplishes of this team have 2–5 Michael E. -

General Glovebox Rules
Glovebox etiquette in the Lewis group Introduction: The gloveboxes are here to assist you in oxygen and water sensitive chemistry. The following guidelines are given to protect the box atmosphere and longevity and ultimately your chemistry and that of the whole group. If you feel that these guidelines impede your ability to do work, talk about it with the person in charge of the glovebox and they will help assist you in designing a set up that will work for you! Disregarding the guidelines compromises your chemistry as well as everyone elses. Available boxes: *Big Bird – Vac, purchased 2008. Drybox, Ar. Noyes 227B. Use for electrochemistry. O2 < 0.5 ppm *Snuffleupagus – Vac, older than time. Drybox, N2. Noyes 227B. Use for electrochemistry and storage. Absolutely no bad solvents/chemicals with the exception of CH3CN open for <5 minutes at a time. O2 < 1 ppm *Oscar the Grouch – Vac, older than time. Flushbox, N2. Noyes 227B. Alcohol OK. Electrochemistry, set-up, surface chemistry. O2 < 10 ppm *Elmo – Vac, purchased 2008. Drybox, N2. BI 032. Electrochemistry and chemical synthesis. O2 < 0.5 ppm *Count von Count – Mbraun, less old than time. Drybox, N2. BI 032. Don, Tony, electrochemistry and set up. O2 < 1 ppm *Cookie Monster – Vac, eh? As old as time?. Flushbox, N2. BI 023. Alcohol OK. Electrochemistry, set-up, surface chemistry. Connected to XPS. O2 < 10 ppm Note: Contact Box Czar before using a new box – even if you are trained on the old box. If nothing else, so they can keep track of who is using what and include you on important info for that box. -

Medical Management of Persons Internally Contaminated with Radionuclides in a Nuclear Or Radiological Emergency
CONTAMINATION EPR-INTERNAL AND RESPONSE PREPAREDNESS EMERGENCY EPR-INTERNAL EPR-INTERNAL CONTAMINATION CONTAMINATION 2018 2018 2018 Medical Management of Persons Internally Contaminated with Radionuclides in a Nuclear or Radiological Emergency Contaminatedin aNuclear with Radionuclides Internally of Persons Management Medical Medical Management of Persons Internally Contaminated with Radionuclides in a Nuclear or Radiological Emergency A Manual for Medical Personnel Jointly sponsored by the Endorsed by AMERICAN SOCIETY FOR RADIATION ONCOLOGY INTERNATIONAL ATOMIC ENERGY AGENCY V I E N N A ISSN 2518–685X @ IAEA SAFETY STANDARDS AND RELATED PUBLICATIONS IAEA SAFETY STANDARDS Under the terms of Article III of its Statute, the IAEA is authorized to establish or adopt standards of safety for protection of health and minimization of danger to life and property, and to provide for the application of these standards. The publications by means of which the IAEA establishes standards are issued in the IAEA Safety Standards Series. This series covers nuclear safety, radiation safety, transport safety and waste safety. The publication categories in the series are Safety Fundamentals, Safety Requirements and Safety Guides. Information on the IAEA’s safety standards programme is available on the IAEA Internet site http://www-ns.iaea.org/standards/ The site provides the texts in English of published and draft safety standards. The texts of safety standards issued in Arabic, Chinese, French, Russian and Spanish, the IAEA Safety Glossary and a status report for safety standards under development are also available. For further information, please contact the IAEA at: Vienna International Centre, PO Box 100, 1400 Vienna, Austria. All users of IAEA safety standards are invited to inform the IAEA of experience in their use (e.g. -

Standard Glovebox Workstations
STANDARD GLOVEBOX WORKSTATIONS www.mbraun.com Light Hood *Freezer Flange DN 40 KF Adjustable Shelving Large Antechamber *Analyzers *Small Antechamber Fine Filter Stainless Steel Piping Programmable Logic Controller Glove Ports Gas Purification System Castors for Easy Mobility Vacuum Pump Leveling Feet Foot Switch *LABstar glovebox workstation is pictured with optional features MBRAUN GLOVEBOX WORKSTATION GLOVEBOX MBRAUN Light Hood Touch Screen PLC Controller *Freezer Adjustable Shelving Large Antechamber *Analyzers Flange DN 40 KF Glove Ports *Small Antechamber Stainless Steel Piping Fine Filter Gas Purification System Castors for Easy Mobility Leveling feet Foot Switch Flow Meter *UNIlab glovebox workstation is pictured with optional features Light Hood Color Touch Screen PLC Controller Adjustable Shelving Large Antechamber Glove Ports *Small Antechamber Fine Filter Flange DN 40 KF Gas Purification Single (SP) or Double (DP) Purifier Unit *External Solvent Trap Castors for Easy Mobility Vacuum Pump Foot Switch Leveling Feet *LABmaster SP/DP glovebox workstation is pictured with optional features GLOVEBOX CONFIGURATIONS MAY VARY LABstar Standard Features: • Programmable logic controller • Large main antechamber • Three glovebox sizes available LAB • Vacuum pump • Automatic regenerable H O/O WORKSTATION 2 2 single column inert gas purifier • World-wide operation using standard power supply The LABstar workstation is a ready to operate high • Integrated high vacuum feedthroughs • Stainless steel adjustable shelving quality glovebox system -

LC-1 Stand Alone Glovebox Operation Manual
LC-1 Stand Alone Glovebox Operation Manual Table of Contents 1. System Overview Page 4 2. Installation Instructions Page 5 - 8 2.1 Attach Gloves Page 5 2.2 Glovebox Connections Page 6 - 8 3. Operational Instructions Page 9 - 39 3.1 Purge System Page 9 - 13 3.1.1 Purging without a Purge Valve Page 9 - 10 3.1.2 Purging with a Manual Purge Valve Page 11 - 12 3.1.3 Purging with an Automatic Purge Valve Page 13 3.2 Normal / Circulation Mode Page 14 - 15 3.3 Antechamber Operation Page 16 - 25 3.3.1 Antechamber Door Operation for Systems Without an Page 16 -17 Antechamber 3.3.2 Antechamber Door Operation for Systems With an Page 18 - 21 Antechamber 3.3.2.1 Bringing Items into Glovebox Page 18 - 20 3.3.2.2 Removing Items from Glovebox Page 21 3.3.3 Mini Antechamber Operation Page 22 - 23 3.3.4 Automatic Antechamber Control Page 24 - 25 3.4 Regeneration Mode Page 26 - 27 3.5 Service Mode Page 28 - 32 3.6 Analyzers Page 33 3.7A Manual Solvent Removal System Operation & Maintenance Page 34 - 35 3.7.1A Manual Solvent Removal System Operation Page 34 3.7.2A Manual Solvent Removal System Maintenance Page 35 3.7B Automatic Solvent Removal System Operation Page 36 - 37 3.7.1B Automatic Solvent Removal System Operation Page 36 3.7.2B Automatic Solvent Removal Reactivation Page 37 3.8 Freezer Operation and Maintenance Page 38 - 39 3.8.1 Freezer Operation Page 38 3.8.2 Freezer Maintenance Page 39 3.9 Box Cooling Operation Page 40 2 3.10 Alarm Messages Page 41 - 42 3.11 Window Removal Page 43 3.12 Window Replacement Page 44 3.13 Maintenance Schedule & Recommended Spare Parts Page 45 3 LC-1 Stand Alone Glovebox Operation Manual Section 1: System Overview Antechamber Star Knobs Antechamber Evacuate/Refill Buttons -35°C Freezer Vacuum Gauge Mini Antechamber Window Frame Mini Antechamber Evacuate/Refill Valve Glove Ports PLC Controller Chiller Butyl Gloves Electrical Cabinet Foot Pedals Gas Purification System Power Switch Solvent Removal System Blower Vacuum Filter Pump Column 4 Section 2: Installation Instructions 2.1 Attach Gloves -Place glove onto the glove port. -

Standard Operating Procedures Standard Operating Procedures Tolman Laboratory
Tolman Group Standard Operating Procedures Standard Operating Procedures Tolman Laboratory v.7 updated May 2016 The intent of this manual is to inform you, the chemist, of the potential hazards that exist in the laboratory. This is by no means a manual to memorize or a manual of instruction. These procedures are not intended to be exhaustive or to serve as a single method of training. The procedures outlined here are meant as a refresher for those who have already received training by senior lab members. If in doubt of any procedure, please ask someone before carrying out said procedure. Asking for help when in doubt is the responsibility of every lab member. The first time you use all equipment and perform new experimental procedures, you should have someone work with you to teach you and to inform you of potential hazards, and to help prevent accidents. The purpose of this manual is to supplement personal one-on-one training with safety information that will protect you personally, as well as other lab members. It is important to make sure that you minimize your exposure to chemicals and decrease the chances of any accidents. By reviewing (and later referring to) this manual, you can be aware of the potential hazards in the lab and learn careful techniques. It is your responsibility to learn about safety and proper handling of chemicals from the resources available, including your laboratory members, other persons in the department, and the Chemical Hygiene Plan, which can be accessed via the web, from the Safety link on the Chemistry Webpage www.chem.umn.edu, or http://www.chem.umn.edu/services/safety/. -

D4.1 Model Materials Experiments: First Dissolution Results
Ref. Ares(2021)285855 - 13/01/2021 Co-funded by the DISCO Grant Agreement: 755443 DELIVERABLE D4.1 Model materials experiments: First dissolution results. Dirk Bosbach (FZJ), C. Cachoir (SCK•CEN), Emmi Myllykylä (VTT), Christophe Jegou (CEA), Joaquin Cobos (CIEMAT), Ian Farnan (UCAM), Claire Corkhill (USFD) Date of issue of this report: 28/06/2019 Report number of pages: 33 p Start date of project: 01/06/2017 Duration: 48 Months Project co-funded by the European Commission under the Euratom Research and Training Programme on Nuclear Energy within the Horizon 2020 Framework Programme Dissemination Level PU Public X PP Restricted to other programme participants (including the Commission Services) RE Restricted to a group specified by the partners of the Disco project CO Confidential, only for partners of the Disco project 1 Introduction The efforts to improve fuel performance in nuclear power generation lead to an increased usage of a various new types of light-water reactor (LWR) fuels including Cr-, Al-, and Si- doped fuels (CEA, 2009, Arborelius, 2006, Massih, 2014). While the improvement of in- reactor performance of these fuels has already been demonstrated, it is still uncertain, whether the corrosion/dissolution behaviour of such fuels in a deep geological repository is similar to conventional spent LWR-fuels. However, the chemical and structural complexity of spent nuclear fuels (SNF) and the associated high beta- and gamma radiation fields impede unravelling all the various concurring dissolution mechanisms from experiments with SNF. Thus within the DisCo project (www.disco-h2020.eu), experiments on irradiated doped fuels are complemented with dissolution studies using systematically produced and carefully characterised UO2-based model materials to understand the effects of the addition of Cr- or Al-oxide into the fuel matrix on SNF dissolution behaviour under repository relevant conditions. -

Electronic Supplementary Information (ESI)
Electronic Supplementary Material (ESI) for Journal of Materials Chemistry A. This journal is © The Royal Society of Chemistry 2016 Electronic Supplementary Information (ESI) Parallel Bulk Heterojunction Photovoltaics Based on All-Conjugated Block Copolymer Additives Jorge W. Mok,a Dylan Kipp,b Luis R. Hasbun,a Andrei Dolocanc, Joseph Strzalka,d Venkat Ganesan,*b and Rafael Verduzco*ae aDepartment of Chemical and Biomolecular Engineering, Rice University, Houston, Texas 77005, United States b Department of Chemical Engineering, University of Texas at Austin, Austin, Texas 78712, United States c Department of Materials Science and Texas Materials Institute, University of Texas at Austin, Austin, Texas 78712, United States dX-ray Science Division, Advanced Photon Source, Argonne National Laboratory, Argonne, Illinois 60439, United States eDepartment of Materials Science and NanoEngineering, Rice University, Houston, Texas 77005, United States *corresponding authors: [email protected] [email protected] Organic solar cells fabrication and measurement: PCBM (C60PXBM, p/n PCBM61-X01) was obtained from Nano-C. The ZnO precursor solution was prepared by dissolving 1 g of zinc acetate dihydrate (≥98%, Sigma-Aldrich) in 10 mL of 2- methoxyethanol (≥99.0%, 50ppm BHT as stabilizer, Sigma-Aldrich) with 0.28 g of ethanolamine (≥99%, Alfa Aesar) as a surfactant. The solution was stirred overnight before use.The PEDOT:PSS solution was prepared by diluting 1 mL of PEDOT:PSS Clevios P VP AI4083 (Heraeus) in 10mL of isopropyl alcohol. ITO glass substrate was cleaned in sequential 15-minute ultrasonic baths using deionized soap water (0.5% Hellmanex III, Helma), deionized water, acetone, and isopropyl alcohol, and dried in an oven at 100˚C for 1 hour. -
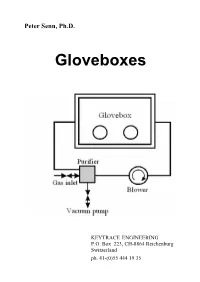
Paper on Gloveboxes
Peter Senn, Ph.D. Gloveboxes KEYTRACE ENGINEERING P.O. Box 223, CH-8864 Reichenburg Switzerland ph. 41-(0)55 444 19 35 Peter Senn, Ph.D. Gloveboxes First Edition US$ 15.- plus postage 1997 Keytrace Engineering ISBN 3-906540-00-6 ABSTRACT Current trends in the design and use of containment workstations or simply "gloveboxes" are discussed. Various designs of commercial glovebox systems are discussed with comments which will put them into perspective for potential applications. Criteria for the assessment and the appropriate design of containment workstations are provided, especially for workstations with recirculatory inert atmosphere systems for handling air- sensitive materials and anaerobic micro-organisms. A simple but fairly realistic mathematical model of gloveboxes with recirculatory gas purification systems is presented. It is shown, that the performance of containment work stations with gas purification systems can be predicted fairly accurately from a performance index , which is inversely proportional to the average time a particle of impurity spends inside the box before being adsorbed. Interactions among gloveboxes in glovebox lines with a single recirculatory gas purification system are also analyzed within the confines of the proposed mathematical model. The performance of protective enclosures with controlled atmospheres might be substantially improved in the foreseeable future by using highly efficient gas purification systems in conjunction with laminar flow techniques similar to the ones used in systems with particulate filters. CONTENTS Chapter Title Page 1. Introduction 1 2. Putting a Glovebox into Operation 6 3. Criteria for Performance Evaluations of Gloveboxes with Inert Atmospheres 10 4. Adsorption of Gaseous Impurities by Reactive Materials 11 5. -

Glovebox General Use
Glovebox General Use Glovebox: Enter and exit the box slowly: The box is not perfect, there are small holes that let ambient atmosphere into the box. Therefore, the box should always be kept under a positive pressure, i.e. not ripping your arms out of the gloves and dropping the glove box pressure lower than atmospheric pressure. If this happens too quickly, and the box cannot compensate quickly enough, it is possible that ambient air could enter the box under the reduced pressure (through the small holes), therefore raising oxygen levels. Antechamber: Evacuate the Antechamber: The antechamber is to be kept under vacuum at all times (except for putting things in the antechamber or removing them from the antechamber). This is standard practice for most antechambers fro the following reasons: (1) It requires the glovebox user to refill it prior to opening the door as a defense against moments of carelessness and (2) it prevents leakage of outside air into the glovebox. Evacuating the antechamber should be the first thing you do after closing the antechamber door. IT SHOULD BE A HABIT! It is orders of magnitude better to accidentally evacuate the antechamber and have to waste a little nitrogen refilling it than it is to forget to evacuate the antechamber. Even if you are coming back out of the box in 5 minutes, EVACUATE THE ANTECHAMBER! Reduce the amount of time the antechamber is left open by having all items you wish to bring in the box ready. Short Cycles are Inexcusable: As all of you should be aware, we use three 10-minute cycles on the small antichamber and three 15-minute cycles on the large antechamber to pump materials into the box, only refilling the chamber halfway (i.e. -

Glovebox Maintainance
Section 5.22 Title: Glovebox Maintenance Revision Date: 11/01/19 Prepared By: Sungho Park and Michael Roy P.I.: Prof. John F. Berry Prior Approval: This procedure is NOT considered hazardous enough that prior approval is needed from the Principal Investigator. Involves Use of Particularly Hazardous Substance (PHS)? No ___ Carcinogen ___ Reproductive Toxin ___ High Acute Toxicity Does this procedure require medical surveillance? No Does this require use of a fit-tested respirator? No Brief Description of Procedure: Maintenance of the gloveboxes. Location: List the locations (buildings/rooms) where this procedure may be performed. For use of a PHS indicate a more precise location within the room, if appropriate, as a designated area. 6319 ("new" double glovebox), 6325 ("old" glovebox), 6375 ("oldest" glovebox) Chemicals Involved: Chemical Physical or Health Hazard (e.g. carcinogen, corrosive) Copper catalyst Inhalation hazard Molecular sieves Inhalation hazard Activated charcoal Inhalation hazard Other Hazards: Include hazards, other than chemical, that may be present during operation of the procedure. High pressure gas cylinders (see gas cylinder SOP) Exposure Controls: (Check all that apply) PPE: _X_ Safety Glasses ___ Face Shield ___Chemical Splash Goggles ___Chemical Apron _X_ Gloves (Nitrile) _X_ Lab Coat ___Respirator (type) _X_ Other: Long sleeves and fabric gloves; Dust mask when replacing charcoal or catalyst bed Engineering Controls: ___ Fume Hood ___Biosafety Cabinet ___ Glove box ___ Vented gas cabinet ___Other: Administrative Controls: List any specific work practices needed to perform this procedure (e.g., cannot be performed alone, must notify other staff members before beginning, etc.). Receive training before performing maintenance. Gas cylinders must always be secured with a chain and belt, both tightened around the cylinder circumference.