United States Patent Office Patented Feb
Total Page:16
File Type:pdf, Size:1020Kb

Load more
Recommended publications
-

Electrolytic Study of Basic Beryllium Acetate in Carbon Disulphide at 303K, 308K, 313K
IARJSET ISSN (Online) 2393-8021 ISSN (Print) 2394-1588 International Advanced Research Journal in Science, Engineering and Technology Vol. 7, Issue 11, November 2020 DOI 10.17148/IARJSET.2020.71106 Electrolytic Study of Basic Beryllium Acetate in Carbon disulphide at 303K, 308K, 313K Rajeev Kumar Sharma1, Renu Singhal2 Assistant Professor, Department of Chemistry, D.S. College, Aligarh, U.P. (India)1 Associate Professor, Department of Chemistry, D.S. College, Aligarh, U.P. (India)2 Abstract: The electrolytic study of Basic Beryllium Acetate (B.B.A.) in carbon disulphide were reported at 303K, 308K, 313K, the solute solvent interaction have been carried out by computing various Physico-chemical Acoustic Parameters [Apparent Molal adiabatic compressibility (k), Isentropic compressibility (S), Intermolecular free length (Lf), Specific Acoustic Impedance (Z), Relative Association and Solvation Number (Sn)], these parameters have been evaluated by using ultrasonic velocity, density, viscosity data. The results of these parameters indicates the strength of molecular interaction. Keywords: Molecular Interaction, Electrolytic, viscous behavior of Basic Beryllium acetate. I. INTRODUCTION The Isentropic compressibility of dilute aqueous solution of electrolytes decreases with increasing conc. indicating a strong interaction of the dissolved ion with the alkanols1-7, the non-aqueous solution of electrolytes have drawn the attention of same workers.7-9 Now, we are reporting a study of Ultrasound velocity, density and viscosity measurement at 300C, 350C, 400C have 8 been used to calculate isentropic compressibility (s), Intermolecular Free Lenght (Lf) , Specific Acoustic Impedance 9 10 (Z) , Molar sound velocity (R), Relative association (RA) , Apparent molal adiabatic compressibility (k), Wada 11 0 0 constant (B), Shear’s relaxation time (s) and Solvation numbers (Sn) of B.B.A. -

Methanogenesis Rates in Acetate and Nitrate Amended Anoxic Slurries
Methanogenesis rates in acetate and nitrate amended anoxic slurries BIOS 35502: Practicum in Environmental Field Biology Patrick Revord Advisor: William West 2011 Abstract With increasing urbanization and land use changes, pollution of lakes and wetland ecosystems is imminent. Any influx of nutrients, anthropogenic or natural, can have dramatic effects on lake gas production and flux. However, the net effect of simultaneous increase of both acetate and nitrate is unknown. Methane (CH4) production was measured in anoxic sediment and water slurries amended with ammonium nitrate (NH4NO3), which has been shown to inhibit methanogenesis, and sodium acetate (CH3COONa or NaOAc), which is known to increase methanogenesis. The addition of acetate significantly increased the methanogenesis rate, but the nitrate amendment had no significant effect. The simultaneous amendment of both acetate and nitrate showed no significant increase in CH4 compared to the control, indicating that the presence of nitrate may have reduced the effect of acetate amendment. Introduction Methane, a greenhouse gas associated with global warming, continues to increase in concentration in our atmosphere. Global yearly flux of methane into the atmosphere is 566 teragrams of CH4 per year, which is more than double pre-industrial yearly flux (Solomon et al. 2007). Increasing urbanization and land-use changes contribute significantly to increased gas levels (Anderson et al. 2010, Vitousek 1994). Nutrients travel from anthropogenic sources such as wastewater treatment facilities, landfills, and agricultural plots into nearby lakes, rivers, and wetlands, causing increased primary productivity in a process known as eutrophication (Vitousek et al. 1997). The increased nutrients and productivity lead to toxic algal blooms that create products such as acetate, H2, and CO2; a nutrient-rich anoxic environment suitable for anaerobic bacteria to produce unnaturally high levels of methane and other greenhouse gases (Davis and Koop 2006, West unpublished data). -

Isoamyl Acetate
SUMMARY OF DATA FOR CHEMICAL SELECTION Isoamyl Acetate CAS No. 123-92-2 Prepared for NTP by Technical Resources International, Inc Prepared on 11/94 Under NCI Contract No. N01-CP-56019 Table of Contents I. Chemical Identification II. Exposure Information Table 1. Levels of isoamyl acetate reported in foods III. Evidence for Possible Carcinogenic Activity Appendix A: Structural Analogs of Isoamyl Acetate IV. References SUMMARY OF DATA FOR CHEMICAL SELECTION CHEMICAL IDENTIFICATION CAS Registry No.: 123-92-2 Chem. Abstr. Name: 1-Butanol, 3-methyl-, acetate Synonyms: Acetic acid 3-methylbutyl ester; acetic acid, isopentyl ester; AI3-00576; banana oil; isoamyl ethanoate; isopentyl acetate; isopentyl alcohol, acetate; pear oil; 3-methyl-1-butanol acetate; 3-methyl-1-butyl acetate; 3-methylbutyl acetate; 3-methylbutyl ethanoate; i-amyl acetate Structure: Molecular Formula and Molecular Weight: C7H14O2 Mol. Wt.: 130.18 Chemical and Physical Properties: Description: Colorless, flammable liquid with a banana-like odor (ACGIH, 1993). Boiling Point: 142°C (Lide, 1993) Melting Point: -78.5°C (Mark, et al, 1984; Lide, 1993) Solubility: Soluble in water (2000 mg/L at 25°C) (Howard, 1990); soluble in ethanol, diethyl ether, and acetone (Lide, 1993). Vapor 4.5 mm Hg at 20°C (Howard, 1990) Pressure: Refractive 1.4003 (Lide, 1993) Index: Flash Point: closed cup, 33°C; open cup, 38°:C (Budavari, 1989) Density: 0.876 (Lewis, 1993) Reactivity: Thermal decomposition of isoamyl acetate may produce acrid fumes. Contact with strong oxidizing agents, strong acids, and alkaline materials should be avoided (Haarmann & Reimer Corp., 1994). Hazardous decomposition products of isoamyl acetate include CO and CO2 (AESAR/Alfa, 1994) Log 2.13 (Howard, 1990) P(octanol/water partition coefficient): Technical Isoamyl acetate is commercially available as both a natural and synthetic product with a purity Products and range of 95-99+%. -

Bond Distances and Bond Orders in Binuclear Metal Complexes of the First Row Transition Metals Titanium Through Zinc
Metal-Metal (MM) Bond Distances and Bond Orders in Binuclear Metal Complexes of the First Row Transition Metals Titanium Through Zinc Richard H. Duncan Lyngdoh*,a, Henry F. Schaefer III*,b and R. Bruce King*,b a Department of Chemistry, North-Eastern Hill University, Shillong 793022, India B Centre for Computational Quantum Chemistry, University of Georgia, Athens GA 30602 ABSTRACT: This survey of metal-metal (MM) bond distances in binuclear complexes of the first row 3d-block elements reviews experimental and computational research on a wide range of such systems. The metals surveyed are titanium, vanadium, chromium, manganese, iron, cobalt, nickel, copper, and zinc, representing the only comprehensive presentation of such results to date. Factors impacting MM bond lengths that are discussed here include (a) n+ the formal MM bond order, (b) size of the metal ion present in the bimetallic core (M2) , (c) the metal oxidation state, (d) effects of ligand basicity, coordination mode and number, and (e) steric effects of bulky ligands. Correlations between experimental and computational findings are examined wherever possible, often yielding good agreement for MM bond lengths. The formal bond order provides a key basis for assessing experimental and computationally derived MM bond lengths. The effects of change in the metal upon MM bond length ranges in binuclear complexes suggest trends for single, double, triple, and quadruple MM bonds which are related to the available information on metal atomic radii. It emerges that while specific factors for a limited range of complexes are found to have their expected impact in many cases, the assessment of the net effect of these factors is challenging. -

NMR Chemical Shifts of Common Laboratory Solvents As Trace Impurities
7512 J. Org. Chem. 1997, 62, 7512-7515 NMR Chemical Shifts of Common Laboratory Solvents as Trace Impurities Hugo E. Gottlieb,* Vadim Kotlyar, and Abraham Nudelman* Department of Chemistry, Bar-Ilan University, Ramat-Gan 52900, Israel Received June 27, 1997 In the course of the routine use of NMR as an aid for organic chemistry, a day-to-day problem is the identifica- tion of signals deriving from common contaminants (water, solvents, stabilizers, oils) in less-than-analyti- cally-pure samples. This data may be available in the literature, but the time involved in searching for it may be considerable. Another issue is the concentration dependence of chemical shifts (especially 1H); results obtained two or three decades ago usually refer to much Figure 1. Chemical shift of HDO as a function of tempera- more concentrated samples, and run at lower magnetic ture. fields, than today’s practice. 1 13 We therefore decided to collect H and C chemical dependent (vide infra). Also, any potential hydrogen- shifts of what are, in our experience, the most popular bond acceptor will tend to shift the water signal down- “extra peaks” in a variety of commonly used NMR field; this is particularly true for nonpolar solvents. In solvents, in the hope that this will be of assistance to contrast, in e.g. DMSO the water is already strongly the practicing chemist. hydrogen-bonded to the solvent, and solutes have only a negligible effect on its chemical shift. This is also true Experimental Section for D2O; the chemical shift of the residual HDO is very NMR spectra were taken in a Bruker DPX-300 instrument temperature-dependent (vide infra) but, maybe counter- (300.1 and 75.5 MHz for 1H and 13C, respectively). -

Ion Chromatographic Analysis of Disinfectant By-Products
DCU Ion Chromatographic Analysis of Disinfectant By-Products For the award of D octor of Philosophy Leon Barron School of Chem ical Sciences Student ID: 97311006 V S > N C S R National Centra for Sansor Raiaireh Table of Contents Declaration 11 List of Figures 12 List of Tables 20 List of Abbreviations 22 List of Publications 23 List of Poster Presentations 24 List of Oral Presentations 25 List of Honours and A wards 2 5 Acknowledgements 26 Abstract 28 Chapter 1.0 Literature Review 30 1.0 Background 31 1.1 Current Water Treatment Methods 32 1.1.1 Ozonation 32 1.1.2 Chlorination 35 1.2 Factors Affecting the Formation of Disinfectant By- 36 Products 1.2.1 pH 36 1.2.2 Contact Time 37 1.2.3 Temperature and Season 37 1.2.4 Concentration and Properties of Natural Organic Matter 37 1.2.5 Concentration of Chlorine and Residual Chlorine 38 1.2.6 Concentration of Bromide 38 2 1.3 The Haloacetic Acids 39 1.3.1 Chlorinated Acetic Acids 39 1.3.1.1 Physical and Chemical Properties 39 1.3.1.2 Effects on Laboratory Animals and/or 39 Humans 1.3.2 Brominated Acetic Acids 40 1.3.2.1 Physical Properties 40 1.3.2.2 Effect on Laboratory Animals and/or 41 Humans 1.4 Chromatographic Determination of Haloacetic Acids 42 1.4.1 Introduction to chromatographic theory 42 1.4.2 Band broadening in chromatography 45 1.4.2.1 Multiple path lengths 45 1.4.2.3 Molecular diffusion 46 1.4.2.4 Mass transfer 47 1.4.2.5 The van Deemter equation 47 1.5 Ion Exchange Chromatography 48 1.5.1 The Ion Exchange Process 49 1.5.1.1 Cation Exchange 49 1.5.1.2 Anion Exchange 49 1.5.2 Ion -

A New Take on the Aspirin Synthesis Lab: a Multi-Step Organic Synthesis Laboratory for High School Students
A new take on the aspirin synthesis lab: A multi-step organic synthesis laboratory for high school students Michael P. Kowalski Rationale The use of the extracts of various plants for the treatment of pain and fever goes back several centuries. A tea made from the leaves or bark of Willow ( Saliaceae ) was found to be especially effective. By the late 19th Century, the active ingredient in these plant extracts, salicylic acid, had been identified. In 1874, the process for the commercial production of salicylic acid was perfected. The use of salicylic acid as an analgesic became widespread. However, side effects, such as gastric bleeding, made long term use of the drug impossible for many patients. In 1895, Felix Hoffman of the Fredrick Bayer Company found a method to acetylate salicylic acid, producing acetyl salicylic acid. This derivative was found to be as effective as salicylic acid as an analgesic but did not have the side effects associated with salicylic acid. The new drug was named “aspirin”, a name inspired by Saint Aspirinius, the patron saint of headaches. The introduction of aspirin marks the beginning of the modern pharmaceutical industry. The mechanism as to how aspirin alleviates pain was not elucidated until the 1970’s, when it was found that aspirin inhibits COX 1 and COX 2, enzymes that catalyze the formation of prostaglandins, substances that cause inflammation (1). The synthesis of aspirin, acetylsalicylic acid, is a standard first year organic chemistry laboratory activity at the college level. In most protocols, salicylic acid is reacted with acetic anhydride producing aspirin and acetic acid by acid catalysis: O OH O O OH O O H2SO4 O CH3 + H3C OH OH H C O CH + 3 3 O The reaction is refluxed for 30 minutes, and then cooled. -
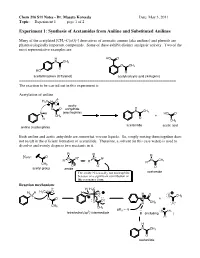
Experiment 1: Synthesis of Acetamides from Aniline and Substituted Anilines
Chem 216 S11 Notes - Dr. Masato Koreeda Date: May 3, 2011 Topic: __Experiment 1____ page 1 of 2. Experiment 1: Synthesis of Acetamides from Aniline and Substituted Anilines Many of the acetylated [CH3–C(=O)-] derivatives of aromatic amines (aka anilines) and phenols are pharmacologically important compounds. Some of these exhibit distinct analgesic activity. Two of the most representative examples are: H HO O N CH3 O CH3 O HO O acetaminophen (Tylenol) acetylsalicylic acid (Aspirin) ======================================================================= The reaction to be carried out in this experiment is: Acetylation of aniline δ- H3C δ+ O acetic O O anhydride H N CH3 N H (electrophile) HO O CH3 + H O CH3 acetanilide acetic acid aniline (nucleophile) Both aniline and acetic anhydride are somewhat viscous liquids. So, simply mixing them together does not result in the efficient formation of acetanilide. Therefore, a solvent (in this case water) is used to dissolve and evenly disperse two reactants in it. R R R Note: O + N R" N R" N CH R' R' R' 3 CH3 - O O O acetyl group amide The amide N is usually not nucleophilic acetamide because of a significant contribution of this resonance form. Reaction mechanism: δ- H H3C H H H3C δ+ O H N O H O N O CH3 O O N + O O H CH3 O CH3 CH3 pKa ~ -5 3 O CH3 tetrahedral (sp ) intermediate B (including ) O H N CH3 O acetanilide Chem 216 S11 Notes - Dr. Masato Koreeda Date: May 3, 2011 Topic: __Experiment 1____ page 2 of 2. Additional comments on the reaction mechanism: 1. -

Aspirin. an Ab Initio Quantum-Mechanical Study of Conformational Preferences and of Neighboring Group Interactions
J. Org. Chem. 2001, 66, 771-779 771 Aspirin. An ab Initio Quantum-Mechanical Study of Conformational Preferences and of Neighboring Group Interactions Rainer Glaser Department of Chemistry, University of MissourisColumbia, Columbia, Missouri 65211 [email protected] Received August 15, 2000 The potential energy surface of acetylsalicylic acid, aspirin, has been explored at the RHF/6-31G* and B3LYP/6-31G* levels, and single-point calculations were performed at levels up to B3LYP/ 6-311G**//B3LYP/6-31G*. All conformational isomers have been located, the thermochemical functions have been computed, and relative energies and free enthalpies were determined. The conformational space of aspirin is spanned by three internal coordinates, and these are the carboxylic acid C-O conformation (s-trans preferred by about 7 kcal/mol), the C-COOH conformation (Z preferred unless there are H-bonding opportunities), and the ester C-O conformation (s-trans preferred by about 4 kcal/mol). There are nine aspirin isomers since one of the conformers realizes hydrogen-bonding structure isomerism as well. Neighboring group interactions are discussed with reference to the intrinsic properties of benzoic acid and phenyl acetate. The intrinsic conformational preference energies for benzoic acid and phenyl acetate are not additive. The acid s-trans preference energies differ by as much as 9 kcal/mol depending on the Ph-COOH and ester conformations. Similarly, the E-preference energies about the Ph-COOH bond vary by as much as 6 kcal/mol depending on the ester conformation. The structural discussion suggests an overall ortho repulsion between the functional groups in all aspirin isomers including the intramolecularly hydrogen- bonded isomers. -

Sodium Acetate
SODIUM ACETATE Prepared at the 18th JECFA (1974), published in NMRS 54B (1975) and in FNP 52 (1992). Metals and arsenic specifications revised at the 59th JECFA (2002). An ADI not limited' was established at the 17th JECFA (1973) SYNONYMS INS No. 262(i) DEFINITION Chemical names Sodium acetate C.A.S. number 127-09-3 Chemical formula C2H3NaO2 · nH2O (n = 0 or 3) Structural formula CH3COONa · nH2O (n = 0 or 3) Formula weight Anhydrous: 82.03 Trihydrate: 136.08 Assay Not less than 98.5% after drying DESCRIPTION Anhydrous: White, odourless, granular, hygroscopic powder Trihydrate: Colourless, transparent crystals or a granular crystalline powder, odourless or with a faint, acetic odour. Effloresces in warm, dry air. FUNCTIONAL USES Buffer CHARACTERISTICS IDENTIFICATION Solubility (Vol. 4) Very soluble in water; soluble in ethanol pH (Vol. 4) 8.0 - 9.5 (1 in 100 soln) Test for sodium (Vol. 4) Passes test Test for acetate (Vol. 4) Passes test Heat test Anhydrous: When heating the sample slowly, it first fuses gradually and boils, and later decomposes evolving an unpleasant odour of acetone. A solution of the residue gives alkaline reaction with litmus paper. Trihydrate: When heating the sample slowly, it liquefies. Then water evaporates, and a powder forms. By heating more strongly, the powder fuses, and becomes lumpy and later decomposes evolving an odour of acetone. A solution of the residue gives alkaline reaction with litmus paper. PURITY Loss on drying (Vol. 4) Anhydrous: Not more than 2.0% (120o, 4 h) Trihydrate: Between 36 and 42% (120o, 4 h) Test for potassium Negative test (Vol. -

Vinyl Acetate
Vinyl acetate 108-05-4 Hazard Summary Vinyl acetate is primarily used as a monomer in the production of polyvinyl acetate and polyvinyl alcohol. Acute (short-term) inhalation exposure of workers to vinyl acetate has resulted in eye irritation and upper respiratory tract irritation. Chronic (long-term) occupational exposure did not result in any severe adverse effects in workers; some instances of upper respiratory tract irritation, cough, and/or hoarseness were reported. Nasal epithelial lesions and irritation and inflammation of the respiratory tract were observed in mice and rats chronically exposed by inhalation. No information is available on the reproductive, developmental, or carcinogenic effects of vinyl acetate in humans. An increased incidence of nasal cavity tumors has been observed in rats exposed by inhalation. In one drinking water study, an increased incidence of tumors was reported in rats. EPA has not classified vinyl acetate for carcinogenicity. Please Note: The main sources of information for this fact sheet are EPA's Integrated Risk Information System (IRIS) (2), which contains information on inhalation chronic toxicity of vinyl acetate and the RfC, and the Agency for Toxic Substances and Disease Registry's (ATSDR's) Toxicological Profile for Vinyl Acetate. (1) Uses Vinyl acetate is primarily used as a monomer in the production of polyvinyl acetate and polyvinyl alcohol.(1) Vinyl acetate is also used as a raw material in the production of other chemicals, in adhesives, water-based paints, nonwoven textile fibers, textile sizings and finishes, paper coatings, inks, films, and lacquers. (1,2) Sources and Potential Exposure Exposure is most likely to occur in the workplace, where individuals may be occupationally exposed to vinyl acetate via inhalation or dermal contact during its manufacture or use. -

The Background Ion Master List Includes
REV. A BACKGROUND ION MASTER LIST 1 OF 5 The background ion master list includes: • ESI+ common background ions on page 1 • ESI- common background ions on page 4 • ESI+ common clusters on page 4 • ESI+ common adducts on page 5 • ESI+ ion series on page 5 Table 1: ESI+ common background ions m/z Ion Compound Source 33 (M+H)+ methanol 42 (M+H)+ acetonitrile + 59 (M+NH4) acetonitrile 64 (M+Na)+ acetonitrile 65 (2M+H)+ methanol 74 (M+H)+ dimethylformamide 79 (M+H)+ DMSO 83 (2M+H)+ acetonitrile 85 (M+H)+ d6-DMSO 88 (M+formic acid+H)+ acetonitrile 101 (M+Na)+ DMSO 102 (M+H)+ triethylamine 104/106 (M+Cu)+ acetonitrile 105 (2M+Na)+ acetonitrile 115 (M+dimethyl formamide+H)+ acetonitrile + 120 (M+Na+CH3CN) DMSO 122 (M+H)+ tris (tris(hydroxymethyl)aminomethane) 123 (M+H)+ diisopropylethylamine 130 (M+H)+ diisopropylethylamine + 137 (M+CH3CN+NH4) DMSO 144 (M+H)+ tripropylamine 145/147 (2M+Cu)+ acetonitrile 146 (3M+Na)+ acetonitrile 149 (M+H)+ phthalic anhydride 150 (M+H)+ phenyldiethylamine 153 (M+H)+ 1,8-diazabicyclo[5.4.0]undec-7-ene (DBU) 157 (2M+H)+ DMSO 159 (M+Na)+ sodium trifluoroacetate 163 dimethyl phthalate 167 dioctyl phthalate 169 (2M+H)+ d6-DMSO 171 (M+Na)+ phthalic anhydride 179 (2M+Na)+ DMSO 186 (M+H)+ tributylamine 195 (M+H)+ dimethyl phthalate 214 (M+H)+ n-butylbenzenesulfonamide plasticizer © 2010 WATERS COPORATION. ALL RIGHTS RESERVED. REV. A BACKGROUND ION MASTER LIST 2 OF 5 Table 1: ESI+ common background ions (continued) m/z Ion Compound Source 225 (M+H)+ dicyclohexylurea + 231 (M+NH4) n-butyl benzenesulfonamide plasticizer