Proteomics, Holm Oak (Quercus Ilex L.) and Other Recalcitrant and Orphan Forest Tree Species: How Do They See Each Other?
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Poplars and Willows: Trees for Society and the Environment / Edited by J.G
Poplars and Willows Trees for Society and the Environment This volume is respectfully dedicated to the memory of Victor Steenackers. Vic, as he was known to his friends, was born in Weelde, Belgium, in 1928. His life was devoted to his family – his wife, Joanna, his 9 children and his 23 grandchildren. His career was devoted to the study and improve- ment of poplars, particularly through poplar breeding. As Director of the Poplar Research Institute at Geraardsbergen, Belgium, he pursued a lifelong scientific interest in poplars and encouraged others to share his passion. As a member of the Executive Committee of the International Poplar Commission for many years, and as its Chair from 1988 to 2000, he was a much-loved mentor and powerful advocate, spreading scientific knowledge of poplars and willows worldwide throughout the many member countries of the IPC. This book is in many ways part of the legacy of Vic Steenackers, many of its contributing authors having learned from his guidance and dedication. Vic Steenackers passed away at Aalst, Belgium, in August 2010, but his work is carried on by others, including mem- bers of his family. Poplars and Willows Trees for Society and the Environment Edited by J.G. Isebrands Environmental Forestry Consultants LLC, New London, Wisconsin, USA and J. Richardson Poplar Council of Canada, Ottawa, Ontario, Canada Published by The Food and Agriculture Organization of the United Nations and CABI CABI is a trading name of CAB International CABI CABI Nosworthy Way 38 Chauncey Street Wallingford Suite 1002 Oxfordshire OX10 8DE Boston, MA 02111 UK USA Tel: +44 (0)1491 832111 Tel: +1 800 552 3083 (toll free) Fax: +44 (0)1491 833508 Tel: +1 (0)617 395 4051 E-mail: [email protected] E-mail: [email protected] Website: www.cabi.org © FAO, 2014 FAO encourages the use, reproduction and dissemination of material in this information product. -

Universita' Degli Studi Di Parma
UNIVERSITA’ DEGLI STUDI DI PARMA DOTTORATO DI RICERCA IN BIOTECNOLOGIE XXIII° CICLO FUNCTIONAL ANALYSIS OF POPLAR (Populus nigra L. and P. nigra x P. deltoids) DURING ENVIRONMENTAL EXPOSURE TO METALS Coordinatore: Chiar.mo Prof. Nelson Marmiroli Relatore: Chiar.mo Prof. Nelson Marmiroli Tutor: Dott.ssa Marta Marmiroli Dott.ssa Giovanna Visioli Dottorando: Dott. Davide Imperiale CONTENTS 1 GENERAL INTRODUCTION .................................................................................................................................. 4 1.1 HEAVY METALS POLLUTION....................................................................................................................................... 4 1.2 REMEDIATION TECHNOLOGIES AND PHYTOREMEDIATION ............................................................................................... 7 1.3 HYPERACCUMULATION .......................................................................................................................................... 10 1.4 MECHANISM OF BIOACCUMULATION IN PLANTS .......................................................................................................... 14 1.5 PHYTOREMEDIATION USING WOODY TREES ................................................................................................................ 17 1.6 PROTEOMICS ....................................................................................................................................................... 21 1.7 PROTEIN SEPARATION TECHNIQUES ......................................................................................................................... -

ANÁLISE in SILICO DE RELAÇÕES FILOGENÉTICAS DE PLANTAS BASEADAS NO GENE Ga20ox
UNIVERSIDADE FEDERAL DO PAMPA CAMPUS SÃO GABRIEL PROGRAMA DE PÓS- GRADUAÇÃO STRICTO SENSU EM CIÊNCIAS BIOLÓGICAS ANÁLISE IN SILICO DE RELAÇÕES FILOGENÉTICAS DE PLANTAS BASEADAS NO GENE GA20ox DISSERTAÇÃO DE MESTRADO LILIAN DE OLIVEIRA MACHADO SÃO GABRIEL, RS, BRASIL 2012 ANÁLISE IN SILICO DE RELAÇÕES FILOGENÉTICAS DE PLANTAS BASEADAS NO GENE GA20ox Lilian de Oliveira Machado Dissertação apresentada ao programa de Pós- Graduação Stricto sensu em Ciências Biológicas da Universidade Federal do Pampa (UNIPAMPA,RS), como requisito parcial para obtenção do Título de Mestre em Ciências Biológicas. Orientador: Prof. Dr. Valdir Marcos Stefenon São Gabriel, RS, Brasil 2012 MACHADO, Lilian de Oliveira Análise In Silico de Relações Filogenética s de Plantas Baseadas no Gene GA20ox / Lilian de Oliveira Machado. 23folhas; Dissertação (Mestrado) Universidade Federal do Pampa, 2012. Orientação: Prof. Dr. Valdir Marcos Stefenon. 1.Sistemática e Ecologia. 2. Ecologia Molecular. 3.Filogenia de plantas. I. Stefenon, Valdir Marcos. II. Doutor Lilian de Oliveira Machado ANÁLISE IN SILICO DE RELAÇÕES FILOGENÉTICAS DE PLANTAS BASEADAS NO GENE GA20ox Dissertação apresentada ao programa de Pós- Graduação Stricto sensu em Ciências Biológicas da Universidade Federal do Pampa (UNIPAMPA,RS), como requisito parcial para obtenção do Título de Mestre em Ciências Biológicas. Área de concentração: Genética. Dissertação defendida e aprovada em 14 de dezembro de 2012. Banca Examinadora: AGRADECIMENTOS Agradeço, primeiramente, a Deus por todas as oportunidades que me ofertou, pela força e coragem nos momentos mais difíceis. Agradeço aos meus queridos pais, Altivo e Saionara, por todo carinho, apoio, compreensão, confiança e amor incondicional. A minha vó amada, Vera!!!! Muito obrigada por tudo que sempre fez por mim, pelo apoio e amor incondicional. -

Intraspecific and Interspecific Genetic and Phylogenetic Relationships In
Theor Appl Genet (2005) 111: 1440–1456 DOI 10.1007/s00122-005-0076-2 ORIGINAL PAPER M. T. Cervera Æ V. Storme Æ A. Soto Æ B. Ivens M. Van Montagu Æ O. P. Rajora Æ W. Boerjan Intraspecific and interspecific genetic and phylogenetic relationships in the genus Populus based on AFLP markers Received: 11 March 2004 / Accepted: 24 June 2005 / Published online: 7 October 2005 Ó Springer-Verlag 2005 Abstract Although Populus has become the model genus pattern consistent with their known evolutionary rela- for molecular genetics and genomics research on forest tionships. A close relationship between Populus deltoides trees, genetic and phylogenetic relationships within this of the Aigeiros section and species of the Tacamahaca genus have not yet been comprehensively studied at the section was observed and, with the exception of Populus molecular level. By using 151 AFLPÒ (AFLPÒ is a wilsonii, between the species of the Leucoides, Taca- registered trademark of Keygene) markers, 178 acces- mahaca, and Aigeiros sections. Populus nigra was clearly sions belonging to 25 poplar species and three inter- separated from its consectional P. deltoides, and should specific hybrids were analyzed, using three accessions be classified separately from P. deltoides. The AFLP belonging to two willow species as outgroups. The ge- profiles pointed out to the lack of divergence between netic and phylogenetic relationships were generally some species and revealed that some accessions corre- consistent with the known taxonomy, although notable sponded with interspecific hybrids. This molecular study exceptions were observed. A dendrogram as well as a provides useful information about genetic relationships single most parsimonious tree, ordered the Populus among several Populus species and, together with mor- sections from the oldest Leuce to the latest Aigeiros,a phological descriptions and crossability, it may help re- view and update systematic classification within the Populus genus. -

Tracheophyte of Xiao Hinggan Ling in China: an Updated Checklist
Biodiversity Data Journal 7: e32306 doi: 10.3897/BDJ.7.e32306 Taxonomic Paper Tracheophyte of Xiao Hinggan Ling in China: an updated checklist Hongfeng Wang‡§, Xueyun Dong , Yi Liu|,¶, Keping Ma | ‡ School of Forestry, Northeast Forestry University, Harbin, China § School of Food Engineering Harbin University, Harbin, China | State Key Laboratory of Vegetation and Environmental Change, Institute of Botany, Chinese Academy of Sciences, Beijing, China ¶ University of Chinese Academy of Sciences, Beijing, China Corresponding author: Hongfeng Wang ([email protected]) Academic editor: Daniele Cicuzza Received: 10 Dec 2018 | Accepted: 03 Mar 2019 | Published: 27 Mar 2019 Citation: Wang H, Dong X, Liu Y, Ma K (2019) Tracheophyte of Xiao Hinggan Ling in China: an updated checklist. Biodiversity Data Journal 7: e32306. https://doi.org/10.3897/BDJ.7.e32306 Abstract Background This paper presents an updated list of tracheophytes of Xiao Hinggan Ling. The list includes 124 families, 503 genera and 1640 species (Containing subspecific units), of which 569 species (Containing subspecific units), 56 genera and 6 families represent first published records for Xiao Hinggan Ling. The aim of the present study is to document an updated checklist by reviewing the existing literature, browsing the website of National Specimen Information Infrastructure and additional data obtained in our research over the past ten years. This paper presents an updated list of tracheophytes of Xiao Hinggan Ling. The list includes 124 families, 503 genera and 1640 species (Containing subspecific units), of which 569 species (Containing subspecific units), 56 genera and 6 families represent first published records for Xiao Hinggan Ling. The aim of the present study is to document an updated checklist by reviewing the existing literature, browsing the website of National Specimen Information Infrastructure and additional data obtained in our research over the past ten years. -
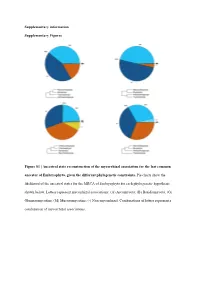
Ancestral State Reconstruction of the Mycorrhizal Association for the Last Common Ancestor of Embryophyta, Given the Different Phylogenetic Constraints
Supplementary information Supplementary Figures Figure S1 | Ancestral state reconstruction of the mycorrhizal association for the last common ancestor of Embryophyta, given the different phylogenetic constraints. Pie charts show the likelihood of the ancestral states for the MRCA of Embryophyta for each phylogenetic hypothesis shown below. Letters represent mycorrhizal associations: (A) Ascomycota; (B) Basidiomycota; (G) Glomeromycotina; (M) Mucoromycotina; (-) Non-mycorrhizal. Combinations of letters represent a combination of mycorrhizal associations. Austrocedrus chilensis Chamaecyparis obtusa Sequoiadendron giganteum Prumnopitys taxifolia Prumnopitys Prumnopitys montana Prumnopitys Prumnopitys ferruginea Prumnopitys Araucaria angustifolia Araucaria Dacrycarpus dacrydioides Dacrycarpus Taxus baccata Podocarpus oleifolius Podocarpus Afrocarpus falcatus Afrocarpus Ephedra fragilis Nymphaea alba Nymphaea Gnetum gnemon Abies alba Abies balsamea Austrobaileya scandens Austrobaileya Abies nordmanniana Thalictrum minus Thalictrum Abies homolepis Caltha palustris Caltha Abies magnifica ia repens Ranunculus Abies religiosa Ranunculus montanus Ranunculus Clematis vitalba Clematis Keteleeria davidiana Anemone patens Anemone Tsuga canadensis Vitis vinifera Vitis Tsuga mertensiana Saxifraga oppositifolia Saxifraga Larix decidua Hypericum maculatum Hypericum Larix gmelinii Phyllanthus calycinus Phyllanthus Larix kaempferi Hieronyma oblonga Hieronyma Pseudotsuga menziesii Salix reinii Salix Picea abies Salix polaris Salix Picea crassifolia Salix herbacea -

A LITERATURE SURVEY of POPULUS SPECIES with EMPHASIS on P. TREMULOIDES Abstract
US. DEPARTMENT OF AGRICULTURE • FOREST SERVICE FOREST PRODUCTS LABORATORY • MADISON, WIS. In cooperation with the University of Wisconsin U. S. D. A. FOREST SERVICE RESEARCH NOTE FPL-0180 (REVISED) AUGUST 1968 A LITERATURE SURVEY OF POPULUS SPECIES WITH EMPHASIS ON P. TREMULOIDES Abstract This revised bibliography reflects the amount of interest in the Populus species shown by the large number of publications issued on the subject, partic ularly in the last two years. References are grouped under broad subject matter headings and arranged alphabetically by author within each group to facilitate use. A LITERATURE SURVEY OF POPULUS SPECIES WITH EMPHASIS ON P. TREMULOIDES By DIMITRI PRONIN and COLEMAN L. VAUGHAN Forest Products Technologists Forest Products Laboratory,1 Forest Service U.S. Department of Agriculture ---- Before 1940, Populus species in the United States were generally considered weed trees. They were seldom utilized, and very little was known about their properties or suitability as a raw material for forest products. This situation has changed dramatically. Large acreages in the Lake States became heavily populated with aspen prior to 1930 as a result of clear-cutting of the original stands of white pine and subsequent severe and repeated fires that burned over the area. In the early 1930's State and Federal agencies improved and expanded their fire detection and suppression systems, which permitted aspen to grow unmolested in vast areas. By the early 1940’s, some of the older aspen stands were reaching merchantable size and timber owners and managers began to search for potential markets. Research organizations and industry launched a concerted effort to determine the potential of aspen as a forest products raw material. -

SALICACEAE 1. POPULUS Linnaeus, Sp. Pl. 2: 1034. 1753
Flora of China 4: 139–274. 1999. 1 SALICACEAE 杨柳科 yang liu ke Fang Zhenfu (方振富 Fang Cheng-fu)1, Zhao Shidong (赵士洞)1; Alexei K. Skvortsov2 Trees or shrubs, deciduous or rarely evergreen, dioecious, rarely polygamous. Leaves alternate, rarely subopposite, usually petiolate, simple; stipules persistent or caducous. Catkins erect or pendulous; each flower usually with a cupular disc or 1 or 2(or 3) nectariferous glands. Male flowers with 2–many stamens; filaments filiform, free or united; to connate; anthers 2(or 4)-loculed, dehiscing longitudinally. Female flowers with 1 pistil, sessile or stipitate; ovary superior, 1- or 2-loculed; ovules several to many, anatropous, with a 1 integument; style 1, 2 in Chosenia; stigmas 2–4. Capsule dehiscing by 2–4(or 5) valves; placenta and inside wall of ovary with long hairs. Seeds 4– numerous, glabrous; hairs and seeds simultaneously deciduous when capsule matures. Three genera and about 620 species: mainly N hemisphere, a few in S hemisphere; three genera and 347 species (236 endemic) in China, including at least nine hybrids and at least one introduced species. Wang Chan & Fang Cheng-fu, eds. 1984. Salicaceae. Fl. Reipubl. Popularis Sin. 20(2): 1–403. 1a. Growth monopodial, buds with several outer scales, terminal bud present (except in Populus sect. Turanga); both male and female catkins pendulous; disc cupular; leaf blade usually 1–2 × as long as wide ......................................................................................................................................................... 1. Populus 1b. Growth sympodial, buds with 1 scale, terminal bud absent; female catkin erect or spreading, very rarely pendulous; flowers without disc but glands sometimes connate and discoid; leaf blade often at least several × as long as wide. -

Forestry Department Food and Agriculture Organization of the United Nations
Forestry Department Food and Agriculture Organization of the United Nations International Poplar Commission Thematic Papers POPLARS AND WILLOWS IN THE WORLD CHAPTER 2 POPLARS AND WILLOWS OF THE WORLD, WITH EMPHASIS ON SILVICULTURALLY IMPORTANT SPECIES Donald I. Dickmann, Julia Kuzovkina October 2008 Forest Resources Development Service Working Paper IPC/9-2 Forest Management Division FAO, Rome, Italy Forestry Department FAO/IPC Poplars and Willows in the World Chapter 2 Poplars and Willows of the World, with Emphasis on Silviculturally Important Species by Donald I. Dickmann Department of Forestry Michigan State University East Lansing, MI 48824-1222 USA Julia Kuzovkina Department of Plant Science The University of Connecticut Storrs, CT 06269-4067 USA Contact information for D. I. Dickmann: Email [email protected] Phone 517 353 5199 Fax 517 432 1143 “…while this planet has gone cycling on according to the fixed laws of gravity, from so simple a beginning endless forms most beautiful and most wonderful have been, and are being, evolved.” Charles Darwin The Origin of Species, 1859 If any family of woody plants affirms Darwin’s musing, it is the Salicaceae. This family—division Magnoliophyta, class Magnoliopsida (dicots), subclass Dilleniidae, order Salicales—includes the familiar genera Populus (poplars, cottonwoods, and aspens) and Salix (willows, sallows, and osiers)1. Together Populus and Salix comprise 400 to 500 species (Table 2-1), although there is no agreement among taxonomists as to the exact number. Added to those numbers are countless subspecies, varieties, hybrids, and cultivars that together encompass a diversity of morphological forms that, although bordering on the incomprehensible, is beautiful and wonderful nonetheless. -
Genome-Wide Comparative Analyses of GATA Transcription Factors Among 19 Arabidopsis Ecotype Genomes: Intraspecifc Characteristics of GATA Transcription Factors
www.nature.com/scientificreports OPEN Genome‑wide comparative analyses of GATA transcription factors among seven Populus genomes Mangi Kim1,2, Hong Xi1,2, Suhyeon Park1,2, Yunho Yun1,2 & Jongsun Park1,2* GATA transcription factors (TFs) are widespread eukaryotic regulators whose DNA‑binding domain is a class IV zinc fnger motif (CX2CX17–20CX2C) followed by a basic region. We identifed 262 GATA genes (389 GATA TFs) from seven Populus genomes using the pipeline of GATA‑TFDB. Alternative splicing forms of Populus GATA genes exhibit dynamics of GATA gene structures including partial or full loss of GATA domain and additional domains. Subfamily III of Populus GATA genes display lack CCT and/ or TIFY domains. 21 Populus GATA gene clusters (PCs) were defned in the phylogenetic tree of GATA domains, suggesting the possibility of subfunctionalization and neofunctionalization. Expression analysis of Populus GATA genes identifed the fve PCs displaying tissue‑specifc expression, providing the clues of their biological functions. Amino acid patterns of Populus GATA motifs display well conserved manner of Populus GATA genes. The fve Populus GATA genes were predicted as membrane‑ bound GATA TFs. Biased chromosomal distributions of GATA genes of three Populus species. Our comparative analysis approaches of the Populus GATA genes will be a cornerstone to understand various plant TF characteristics including evolutionary insights. A transcription factor (TF) is a protein that controls the rate of transcription by binding to specifc DNA sequences, including promoter regions. TF can also combine and interact with cis-acting elements in the pro- moter region as well as interact with other proteins to regulate the start site of transcription1. -

CONSENSUS DOCUMENT on the BIOLOGY of POPULUS L. (POPLARS) English - Or
Unclassified ENV/JM/MONO(2000)10 Organisation de Coopération et de Développement Economiques Organisation for Economic Co-operation and Development 05-Mar-2001 ___________________________________________________________________________________________ English - Or. English ENVIRONMENT DIRECTORATE Unclassified ENV/JM/MONO(2000)10 JOINT MEETING OF THE CHEMICALS COMMITTEE AND THE WORKING PARTY ON CHEMICALS, PESTICIDES AND BIOTECHNOLOGY Series on Harmonization of Regulatory Oversight in Biotechnology No. 16 CONSENSUS DOCUMENT ON THE BIOLOGY OF POPULUS L. (POPLARS) English - Or. English JT00103743 Document complet disponible sur OLIS dans son format d’origine Complete document available on OLIS in its original format ENV/JM/MONO(2000)10 Also published in the Series on Harmonization of Regulatory Oversight in Biotechnology: No. 1, Commercialisation of Agricultural Products Derived through Modern Biotechnology: Survey Results (1995) No. 2, Analysis of Information Elements Used in the Assessment of Certain Products of Modern Biotechnology (1995) No. 3, Report of the OECD Workshop on the Commercialisation of Agricultural Products Derived through Modern Biotechnology (1995) No. 4, Industrial Products of Modern Biotechnology Intended for Release to the Environment: The Proceedings of the Fribourg Workshop (1996) No. 5, Consensus Document on General Information concerning the Biosafety of Crop Plants Made Virus Resistant through Coat Protein Gene-Mediated Protection (1996) No. 6, Consensus Document on Information Used in the Assessment of Environmental Applications Involving Pseudomonas (1997) No. 7, Consensus Document on the Biology of Brassica napus L. (Oilseed Rape) (1997) No. 8, Consensus Document on the Biology of Solanum tuberosum subsp. tuberosum (Potato) (1997) No. 9, Consensus Document on the Biology of Triticum aestivum (Bread Wheat) (1999) No. 10, Consensus Document on General Information Concerning the Genes and Their Enzymes that Confer Tolerance to Glyphosate Herbicide (1999) No. -

The Flora of the Former £Íczyca Iron Ore Dumps
Biodiv. Res. Conserv. 13: 31-36, 2009 BRC www.brc.amu.edu.pl 10.2478/v10119-009-0006-2 The flora of the former £Íczyca iron ore dumps Jan T. SiciÒski1 & Jaros≥aw Sieradzki2 1Department of Geobotany and Plant Ecology, University of £Ûdü, Banacha 12/16, 90-237 £Ûdü, Poland, e-mail: [email protected] 2Departament of Nature Conservation, University of £Ûdü, Banacha 1/3, 90-237 £Ûdü, Poland, e-mail: [email protected] Abstract: Iron ore mines of the £Íczyca region started operating in the sixties and seventies of the 20th century and brought about considerable changes in the agricultural landscape of the town of £Íczyca and its vicinity. As a result of the ore exploitation, rock dumps as well as local depressions were created there. The subject of the present research was flora occurring in areas transformed by iron ore mining in the £Íczyca region. The flora is of spontaneous and anthropogenic (as a result of planting ñ reclamation) character. The results of the conducted research indicate that areas transformed by mining significantly increase their biological diversity. Key words: ore mine dumps, flora, £Íczyca, Middle Poland 1. Introduction Geological drilling carried out at the town of £Íczyca and its vicinities in 1953-1954 testified to the occurrence of iron ore deposits there. In 1955 a mine of siderite iron ore started operating in the area. The towers of £Íczyca mines and high dumps that then appeared became a new element of the landscape of the town and its surroundings. £Íczyca became a new mining center in Poland.