Molecular Dissection of Neurodevelopmental Disorder-Causing Mutations in CYFIP2
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Systems Analysis Implicates WAVE2&Nbsp
JACC: BASIC TO TRANSLATIONAL SCIENCE VOL.5,NO.4,2020 ª 2020 THE AUTHORS. PUBLISHED BY ELSEVIER ON BEHALF OF THE AMERICAN COLLEGE OF CARDIOLOGY FOUNDATION. THIS IS AN OPEN ACCESS ARTICLE UNDER THE CC BY-NC-ND LICENSE (http://creativecommons.org/licenses/by-nc-nd/4.0/). PRECLINICAL RESEARCH Systems Analysis Implicates WAVE2 Complex in the Pathogenesis of Developmental Left-Sided Obstructive Heart Defects a b b b Jonathan J. Edwards, MD, Andrew D. Rouillard, PHD, Nicolas F. Fernandez, PHD, Zichen Wang, PHD, b c d d Alexander Lachmann, PHD, Sunita S. Shankaran, PHD, Brent W. Bisgrove, PHD, Bradley Demarest, MS, e f g h Nahid Turan, PHD, Deepak Srivastava, MD, Daniel Bernstein, MD, John Deanfield, MD, h i j k Alessandro Giardini, MD, PHD, George Porter, MD, PHD, Richard Kim, MD, Amy E. Roberts, MD, k l m m,n Jane W. Newburger, MD, MPH, Elizabeth Goldmuntz, MD, Martina Brueckner, MD, Richard P. Lifton, MD, PHD, o,p,q r,s t d Christine E. Seidman, MD, Wendy K. Chung, MD, PHD, Martin Tristani-Firouzi, MD, H. Joseph Yost, PHD, b u,v Avi Ma’ayan, PHD, Bruce D. Gelb, MD VISUAL ABSTRACT Edwards, J.J. et al. J Am Coll Cardiol Basic Trans Science. 2020;5(4):376–86. ISSN 2452-302X https://doi.org/10.1016/j.jacbts.2020.01.012 JACC: BASIC TO TRANSLATIONALSCIENCEVOL.5,NO.4,2020 Edwards et al. 377 APRIL 2020:376– 86 WAVE2 Complex in LVOTO HIGHLIGHTS ABBREVIATIONS AND ACRONYMS Combining CHD phenotype–driven gene set enrichment and CRISPR knockdown screening in zebrafish is an effective approach to identifying novel CHD genes. -

Encapsulation of the Cytoskeleton: Towards Mimicking the Mechanics of a Cell
! ! ! ! ! "#$%&'()%*+,#!,-!*./!$0*,'1/)/*,#2!*,3%45'!6+6+$1+#7!*./!6/$.%#+$'!,-!%!$/))! ! #$%&$'!($%&)'*$+,&$-!.//,0!12!3)4!$-5-6-+! ! $!7,8$'9:,09!;<!=,6&$0)6$/!>0?)0,,')0?-!@0)A,'%)9B!;<!=)6&)?$0-!.00!.'5;'-!=)6&)?$0-!@C.!! 5!7,8$'9:,09!;<!();:,+)6$/!>0?)0,,')0?-!@0)A,'%)9B!;<!=)6&)?$0-!.00!.'5;'-!=)6&)?$0-!@C.!! 6!D,//4/$'!$0+!=;/,64/$'!();/;?B!1';?'$:-!@0)A,'%)9B!;<!=)6&)?$0-!.00!.'5;'-!=)6&)?$0-!@C.!! +!7,8$'9:,09!;<!();8&B%)6%-!@0)A,'%)9B!;<!=)6&)?$0-!.00!.'5;'-!=)6&)?$0-!@C.!! ! D;'',%8;0+)0?!$49&;'E! .2123E!$//,0/)4F4:)6&2,+4G!HIJK!L$BM$'+!C9',,9-!@0)A,'%)9B!;<!=)6&)?$0-!.00!.'5;'-!=)6&)?$0! NO"KP2!Q,/E!R"!SINTSUNTSS"P2! ! ! ! "! ! 89'*4%$*! Q&,!6B9;%V,/,9;0!;<!$!6,//!6;09';/%!$//!9&,!$%8,69%!;<!6,//!%&$8,!6&$0?,%!$0+!:;9)/)9B!<';:!)9%! 8&B%);/;?)6$/!<4069);0%!<;'!%4'A)A$/!9;!',8';+469);0!9;!+,$9&2!Q&,!%9'4694',!$0+!+B0$:)6%!;<!9&,! 6B9;%V,/,9$/! 6;:8;0,09%E! $69)0-! :)6';9454/,%-! )09,':,+)$9,! <)/$:,09%-! $0+! %,89)0%! T! ',6,09/B! ',?$'+,+!$%!9&,!<;4'9&!:,:5,'!;<!9&,!6B9;%V,/,9;0!<$:)/B!T!$',!6;0%,'A,+!+4')0?!,A;/49);02!C46&! 6;0%,'A,+! $0+! ,<<,69)A,! 6;09';/! ;A,'! 9&,! :,6&$0)6%! ;<! 9&,! 6,//! :$V,%! 9&,! 6B9;%V,/,9$/! 6;:8;0,09%!?',$9!6$0+)+$9,%!<;'!!"#$!%&'!',6;0%9)949);0!$0+!5;99;:T48!%B09&,9)6!5);/;?B!%94+),%2! L,',-!M,!',A),M!9&,!',6,09!,<<;'9%!)0!',6;0%9)949);0!;<!9&,!6B9;%V,/,9;0!)0!$0+!;0!:,:5'$0,T ,06/;%,+!5);:):,9)6!%B%9,:%!$0+!$'?4,!9&$9!6;T',6;0%9)949);0!$0+!%B0,'?)%9)6!)09,'8/$B!5,9M,,0! 6B9;%V,/,9$/! <)/$:,09%! :)?&9! 5,! )0+)%8,0%$5/,! <;'! ,<<)6),09! :,6&$0)6$/! <4069);0$/)9B! ;<! $69)A,! :)0):$/!6,//%2!W4'9&,'-!:,6&$0)6$/!,X4)/)5')4:!)0!$+&,',09!,4V$'B;9)6!6,//%!)%!$6&),A,+!5B!9&,! -

Identification of Novel E2F1 Target Genes Regulated in Cell Cycle
Oncogene (2006) 25, 1786–1798 & 2006 Nature Publishing Group All rights reserved 0950-9232/06 $30.00 www.nature.com/onc ORIGINAL ARTICLE Identification of novel E2F1 target genes regulated in cell cycle-dependent and independent manners R Iwanaga1,3, H Komori1, S Ishida2, N Okamura3, K Nakayama4, KI Nakayama5 and K Ohtani1 1Human Gene Sciences Center, Tokyo Medical and Dental University, Bunkyo-ku, Tokyo, Japan; 2Division of Pharmacology, National Institute of Health Sciences, Setagaya-ku, Tokyo, Japan; 3Laboratory of Microbiology and Immunology, Graduate School of Health Sciences, Tokyo Medical and Dental University, Bunkyo-ku, Tokyo, Japan; 4Department of Developmental Biology, Center for Translational and Advanced Animal Research on Human Disease, Graduate School of Medicine, Tohoku University, Aoba-ku, Sendai, Japan and 5Department of Molecular and Cellular Biology, Medical Institute of Bioregulation, Kyushu University, Fukuoka, Japan The transcription factor E2F mediates cell cycle-depen- cell cycle progression (Nevins et al., 1997; Dyson, 1998; dent expression of genes important for cell proliferation in Trimarchi and Lees, 2002). The transcriptional ability of response to growth stimulation. To further understand the E2F is cell cycle-regulated mainly through association role of E2F, we utilized a sensitive subtraction method to with the retinoblastoma tumor suppressor family of explore new E2F1 targets, which are expressed at low proteins pRb, p107 and p130. During the progression of levels and might have been unrecognized in previous cells from G1 to S phase, G1 cyclin-dependent kinases studies. We identified 33 new E2F1-inducible genes, (cdks)phosphorylate and dissociate the pRb family including checkpoint genes Claspin and Rad51ap1, and proteins from E2F, resulting in the activation of a group four genes with unknown function required for cell cycle of genes required for progression into the S phase. -

Mouse Wasf1 Knockout Project (CRISPR/Cas9)
https://www.alphaknockout.com Mouse Wasf1 Knockout Project (CRISPR/Cas9) Objective: To create a Wasf1 knockout Mouse model (C57BL/6J) by CRISPR/Cas-mediated genome engineering. Strategy summary: The Wasf1 gene (NCBI Reference Sequence: NM_031877 ; Ensembl: ENSMUSG00000019831 ) is located on Mouse chromosome 10. 9 exons are identified, with the ATG start codon in exon 2 and the TAA stop codon in exon 9 (Transcript: ENSMUST00000019975). Exon 3~7 will be selected as target site. Cas9 and gRNA will be co-injected into fertilized eggs for KO Mouse production. The pups will be genotyped by PCR followed by sequencing analysis. Note: Mutation of this gene has been associated with both morphological and functional defects of the central nervous system. Targeted mutagenesis has resulted in mice that display sensorimotor and cognitive defects similar to those exhibited by patients with 3p-syndrome mental retardation. Exon 3 starts from about 7.99% of the coding region. Exon 3~7 covers 45.32% of the coding region. The size of effective KO region: ~8173 bp. The KO region does not have any other known gene. Page 1 of 8 https://www.alphaknockout.com Overview of the Targeting Strategy Wildtype allele 5' gRNA region gRNA region 3' 1 3 4 5 6 7 9 Legends Exon of mouse Wasf1 Knockout region Page 2 of 8 https://www.alphaknockout.com Overview of the Dot Plot (up) Window size: 15 bp Forward Reverse Complement Sequence 12 Note: The 2000 bp section upstream of Exon 3 is aligned with itself to determine if there are tandem repeats. No significant tandem repeat is found in the dot plot matrix. -

A Glance on the Role of Actin in Osteogenic and Adipogenic
Khan et al. Stem Cell Research & Therapy (2020) 11:283 https://doi.org/10.1186/s13287-020-01789-2 REVIEW Open Access A glance on the role of actin in osteogenic and adipogenic differentiation of mesenchymal stem cells Asmat Ullah Khan† , Rongmei Qu† , Tingyu Fan , Jun Ouyang* and Jingxing Dai* Abstract Mesenchymal stem cells (MSCs) have the capacity to differentiate into multiple lineages including osteogenic and adipogenic lineages. An increasing number of studies have indicated that lineage commitment by MSCs is influenced by actin remodeling. Moreover, actin has roles in determining cell shape, nuclear shape, cell spreading, and cell stiffness, which eventually affect cell differentiation. Osteogenic differentiation is promoted in MSCs that exhibit a large spreading area, increased matrix stiffness, higher levels of actin polymerization, and higher density of stress fibers, whereas adipogenic differentiation is prevalent in MSCs with disrupted actin networks. In addition, the mechanical properties of F-actin empower cells to sense and transduce mechanical stimuli, which are also reported to influence differentiation. Various biomaterials, mechanical, and chemical interventions along with pathogen- induced actin alteration in the form of polymerization and depolymerization in MSC differentiation were studied recently. This review will cover the role of actin and its modifications through the use of different methods in inducing osteogenic and adipogenic differentiation. Keywords: Mesenchymal stem cells (MSCs), Actin, Osteogenesis, Adipogenesis cytoskeleton, Osteogenic differentiation, Adipogenic differentiation, Cytoskeleton Introduction however, this review will focus specifically on the role of Stem cells exhibit a great potential for use in tissue en- actin. gineering because of their regenerative capacity in many Actin is a globular protein with a molecular weight of tissues, including nervous tissue, muscle tissue, adipose approximately 42 kDa and consists of four structural do- tissue, cartilage tissue, and bone tissue. -

Identification of Potential Key Genes and Pathway Linked with Sporadic Creutzfeldt-Jakob Disease Based on Integrated Bioinformatics Analyses
medRxiv preprint doi: https://doi.org/10.1101/2020.12.21.20248688; this version posted December 24, 2020. The copyright holder for this preprint (which was not certified by peer review) is the author/funder, who has granted medRxiv a license to display the preprint in perpetuity. All rights reserved. No reuse allowed without permission. Identification of potential key genes and pathway linked with sporadic Creutzfeldt-Jakob disease based on integrated bioinformatics analyses Basavaraj Vastrad1, Chanabasayya Vastrad*2 , Iranna Kotturshetti 1. Department of Biochemistry, Basaveshwar College of Pharmacy, Gadag, Karnataka 582103, India. 2. Biostatistics and Bioinformatics, Chanabasava Nilaya, Bharthinagar, Dharwad 580001, Karanataka, India. 3. Department of Ayurveda, Rajiv Gandhi Education Society`s Ayurvedic Medical College, Ron, Karnataka 562209, India. * Chanabasayya Vastrad [email protected] Ph: +919480073398 Chanabasava Nilaya, Bharthinagar, Dharwad 580001 , Karanataka, India NOTE: This preprint reports new research that has not been certified by peer review and should not be used to guide clinical practice. medRxiv preprint doi: https://doi.org/10.1101/2020.12.21.20248688; this version posted December 24, 2020. The copyright holder for this preprint (which was not certified by peer review) is the author/funder, who has granted medRxiv a license to display the preprint in perpetuity. All rights reserved. No reuse allowed without permission. Abstract Sporadic Creutzfeldt-Jakob disease (sCJD) is neurodegenerative disease also called prion disease linked with poor prognosis. The aim of the current study was to illuminate the underlying molecular mechanisms of sCJD. The mRNA microarray dataset GSE124571 was downloaded from the Gene Expression Omnibus database. Differentially expressed genes (DEGs) were screened. -

Role and Regulation of the Actin-Regulatory Protein Hs1 in Tcr Signaling
University of Pennsylvania ScholarlyCommons Publicly Accessible Penn Dissertations Fall 2009 Role and Regulation of the Actin-Regulatory Protein Hs1 in Tcr Signaling Esteban Carrizosa University of Pennsylvania, [email protected] Follow this and additional works at: https://repository.upenn.edu/edissertations Part of the Cell Biology Commons, and the Immunology and Infectious Disease Commons Recommended Citation Carrizosa, Esteban, "Role and Regulation of the Actin-Regulatory Protein Hs1 in Tcr Signaling" (2009). Publicly Accessible Penn Dissertations. 91. https://repository.upenn.edu/edissertations/91 This paper is posted at ScholarlyCommons. https://repository.upenn.edu/edissertations/91 For more information, please contact [email protected]. Role and Regulation of the Actin-Regulatory Protein Hs1 in Tcr Signaling Abstract Numerous aspects of T cell function, including TCR signaling, migration, and execution of effector functions, depend on the actin cytoskeleton. Cytoskeletal rearrangements are driven by the action of actin-regulatory proteins, which promote or antagonize the assembly of actin filaments in esponser to external cues. In this work, we have examined the regulation and function of HS1, a poorly-understood actin regulatory protein, in T cells. This protein, which becomes tyrosine phosphorylated upon T cell activation, is thought to function primarily by stabilizing existing branched actin filaments. Loss of HS1 results in unstable actin responses upon TCR engagement and defective Ca2+ responses, leading to poor activation of the IL2 promoter. TCR engagement leads to phosphorylation of HS1 at Tyr 378 and Tyr 397, creating binding sites for SH2 domain-containing proteins, including Vav1 and Itk. Phosphorylation at these residues is required for Itk-dependent recruitment of HS1 to the IS, Vav1 IS localization, and HS1-dependent actin reorganization and IL2 production. -

Genomic Selection Signatures in Autism Spectrum Disorder Identifies
www.nature.com/scientificreports OPEN Genomic selection signatures in autism spectrum disorder identifes cognitive genomic tradeof and its relevance in paradoxical phenotypes of defcits versus potentialities Anil Prakash1,2 & Moinak Banerjee1* Autism spectrum disorder (ASD) is a heterogeneous neurodevelopmental disorder characterized by paradoxical phenotypes of defcits as well as gain in brain function. To address this a genomic tradeof hypothesis was tested and followed up with the biological interaction and evolutionary signifcance of positively selected ASD risk genes. SFARI database was used to retrieve the ASD risk genes while for population datasets 1000 genome data was used. Common risk SNPs were subjected to machine learning as well as independent tests for selection, followed by Bayesian analysis to identify the cumulative efect of selection on risk SNPs. Functional implication of these positively selected risk SNPs was assessed and subjected to ontology analysis, pertaining to their interaction and enrichment of biological and cellular functions. This was followed by comparative analysis with the ancient genomes to identify their evolutionary patterns. Our results identifed signifcant positive selection signals in 18 ASD risk SNPs. Functional and ontology analysis indicate the role of biological and cellular processes associated with various brain functions. The core of the biological interaction network constitutes genes for cognition and learning while genes in the periphery of the network had direct or indirect impact on brain function. Ancient genome analysis identifed de novo and conserved evolutionary selection clusters. The de-novo evolutionary cluster represented genes involved in cognitive function. Relative enrichment of the ASD risk SNPs from the respective evolutionary cluster or biological interaction networks may help in addressing the phenotypic diversity in ASD. -
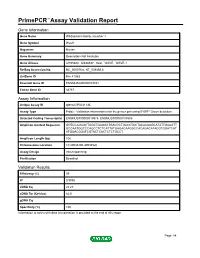
Primepcr™Assay Validation Report
PrimePCR™Assay Validation Report Gene Information Gene Name WAS protein family, member 1 Gene Symbol Wasf1 Organism Mouse Gene Summary Description Not Available Gene Aliases AI195380, AI838537, Scar, WAVE, WAVE-1 RefSeq Accession No. NC_000076.6, NT_039492.8 UniGene ID Mm.41353 Ensembl Gene ID ENSMUSG00000019831 Entrez Gene ID 83767 Assay Information Unique Assay ID qMmuCIP0031135 Assay Type Probe - Validation information is for the primer pair using SYBR® Green detection Detected Coding Transcript(s) ENSMUST00000019975, ENSMUST00000105509 Amplicon Context Sequence GGTCCAGAGCTGGCTGAGGATGACGCTGACCTCCTACACAAGCATATTGAAGTT GCCAATGGCCCAGCCTCTCATTATGAGACAAGGCCACAGACATACGTGGATCAT ATGGACGGATCGTACTCACTCTCTGCCT Amplicon Length (bp) 106 Chromosome Location 10:40933200-40934526 Assay Design Intron-spanning Purification Desalted Validation Results Efficiency (%) 99 R2 0.9998 cDNA Cq 22.23 cDNA Tm (Celsius) 82.5 gDNA Cq Specificity (%) 100 Information to assist with data interpretation is provided at the end of this report. Page 1/4 PrimePCR™Assay Validation Report Wasf1, Mouse Amplification Plot Amplification of cDNA generated from 25 ng of universal reference RNA Melt Peak Melt curve analysis of above amplification Standard Curve Standard curve generated using 20 million copies of template diluted 10-fold to 20 copies Page 2/4 PrimePCR™Assay Validation Report Products used to generate validation data Real-Time PCR Instrument CFX384 Real-Time PCR Detection System Reverse Transcription Reagent iScript™ Advanced cDNA Synthesis Kit for RT-qPCR Real-Time PCR Supermix SsoAdvanced™ SYBR® Green Supermix Experimental Sample qPCR Mouse Reference Total RNA Data Interpretation Unique Assay ID This is a unique identifier that can be used to identify the assay in the literature and online. Detected Coding Transcript(s) This is a list of the Ensembl transcript ID(s) that this assay will detect. -

(SCD) Among Indian Patients Using Gene Expression Data Analysis
www.bioinformation.net Hypothesis Volume 14(7) Identification of therapeutic targets for inflammation in sickle cell disease (SCD) among Indian patients using gene expression data analysis Ipsita Das1, Hrishikesh Mishra2, Prafulla K. Khodiar1, Pradeep K. Patra1* 1Pt. J.N.M. Medical College, Raipur, India; 2Sickle Cell Institute Chhattisgarh, Raipur, India; Pradeep K Patra – E-mail: [email protected]; Phone: 91-771-2890012; *Corresponding author Received July 24, 2018; Revised July 29, 2018; Accepted July 29, 2018; Published July 31, 2018 doi: 10.6026/97320630014408 Abstract: Sickle cell disease (SCD) is life-threatening hemoglobinopathy prevalent in India, Sub-Saharan Africa and Middle East. Inflammation plays a pivotal role in disease process and involves intricate interaction among leukocytes, platelets, sickle erythrocytes and vascular endothelium. Available disease modifying therapies are hydroxyl-urea and blood transfusion. Therefore, it is of interest to develop improved pharmacological agents for SCD. We report up-regulated genes in steady state and vaso-occlusive crisis using analysis of gene expression data obtained by microarray experiment for SCD as potential targets. The association of these targets with inflammation in pathway analysis is also documented. Keywords: Sickle cell disease, vaso-occlusive crisis, inflammation, gene expression, pathophysiology, drug targets. Background: vascular endothelium. Leukocytosis and activation of neutrophils Sickle cell disease (SCD) is a life threatening hemoglobin disorder and monocytes further increases vascular inflammation and affecting about 5% of world population and is prevalent in India endothelial damage and plays as a trigger for VOC [3]. In and other parts of the world including Sub-Saharan Africa and addition to vaso-occlusion, inflammation plays a pivotal role in Middle East. -

Increased CYFIP1 Dosage Alters Cellular and Dendritic Morphology and Dysregulates Mtor
Molecular Psychiatry (2015) 20, 1069–1078 © 2015 Macmillan Publishers Limited All rights reserved 1359-4184/15 www.nature.com/mp ORIGINAL ARTICLE Increased CYFIP1 dosage alters cellular and dendritic morphology and dysregulates mTOR A Oguro-Ando1,2, C Rosensweig1,5, E Herman1,6, Y Nishimura1,7, D Werling1, BR Bill1, JM Berg1, F Gao1, G Coppola1,3, BS Abrahams1,8 and DH Geschwind1,4 Rare maternally inherited duplications at 15q11-13 are observed in ~ 1% of individuals with an autism spectrum disorder (ASD), making it among the most common causes of ASD. 15q11-13 comprises a complex region, and as this copy number variation encompasses many genes, it is important to explore individual genotype–phenotype relationships. Cytoplasmic FMR1-interacting protein 1 (CYFIP1) is of particular interest because of its interaction with Fragile X mental retardation protein (FMRP), its upregulation in transformed lymphoblastoid cell lines from patients with duplications at 15q11-13 and ASD and the presence of smaller overlapping deletions of CYFIP1 in patients with schizophrenia and intellectual disability. Here, we confirm that CYFIP1 is upregulated in transformed lymphoblastoid cell lines and demonstrate its upregulation in the post-mortem brain from 15q11-13 duplication patients for the first time. To investigate how increased CYFIP1 dosage might predispose to neurodevelopmental disease, we studied the consequence of its overexpression in multiple systems. We show that overexpression of CYFIP1 results in morphological abnormalities including cellular hypertrophy in SY5Y cells and differentiated mouse neuronal progenitors. We validate these results in vivo by generating a BAC transgenic mouse, which overexpresses Cyfip1 under the endogenous promotor, observing an increase in the proportion of mature dendritic spines and dendritic spine density. -

Cytoskeletal Remodeling in Cancer
biology Review Cytoskeletal Remodeling in Cancer Jaya Aseervatham Department of Ophthalmology, University of Texas Health Science Center at Houston, Houston, TX 77054, USA; [email protected]; Tel.: +146-9767-0166 Received: 15 October 2020; Accepted: 4 November 2020; Published: 7 November 2020 Simple Summary: Cell migration is an essential process from embryogenesis to cell death. This is tightly regulated by numerous proteins that help in proper functioning of the cell. In diseases like cancer, this process is deregulated and helps in the dissemination of tumor cells from the primary site to secondary sites initiating the process of metastasis. For metastasis to be efficient, cytoskeletal components like actin, myosin, and intermediate filaments and their associated proteins should co-ordinate in an orderly fashion leading to the formation of many cellular protrusions-like lamellipodia and filopodia and invadopodia. Knowledge of this process is the key to control metastasis of cancer cells that leads to death in 90% of the patients. The focus of this review is giving an overall understanding of these process, concentrating on the changes in protein association and regulation and how the tumor cells use it to their advantage. Since the expression of cytoskeletal proteins can be directly related to the degree of malignancy, knowledge about these proteins will provide powerful tools to improve both cancer prognosis and treatment. Abstract: Successful metastasis depends on cell invasion, migration, host immune escape, extravasation, and angiogenesis. The process of cell invasion and migration relies on the dynamic changes taking place in the cytoskeletal components; actin, tubulin and intermediate filaments. This is possible due to the plasticity of the cytoskeleton and coordinated action of all the three, is crucial for the process of metastasis from the primary site.