The Acyl-Coa Thioesterase I (Acot1)
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Inflammatory Stimuli Induce Acyl-Coa Thioesterase 7 and Remodeling of Phospholipids Containing Unsaturated Long (C20)-Acyl Chains in Macrophages
Supplemental Material can be found at: http://www.jlr.org/content/suppl/2017/04/17/jlr.M076489.DC1 .html Inflammatory stimuli induce acyl-CoA thioesterase 7 and remodeling of phospholipids containing unsaturated long (C20)-acyl chains in macrophages Valerie Z. Wall,*,† Shelley Barnhart,* Farah Kramer,* Jenny E. Kanter,* Anuradha Vivekanandan-Giri,§ Subramaniam Pennathur,§ Chiara Bolego,** Jessica M. Ellis,§§,*** Miguel A. Gijón,††† Michael J. Wolfgang,*** and Karin E. Bornfeldt1,*,† Department of Medicine,* Division of Metabolism, Endocrinology and Nutrition, and Department of Pathology,† UW Medicine Diabetes Institute, University of Washington, Seattle, WA; Department of Internal Medicine,§ University of Michigan, Ann Arbor, MI; Department of Pharmaceutical and Pharmacological Sciences,** University of Padova, Padova, Italy; Department of Nutrition Science,§§ Purdue University, West Lafayette, IN; Department of Biological Chemistry,*** Johns Hopkins University School of Medicine, Baltimore, MD; and Department of Pharmacology,††† University of Downloaded from Colorado Denver, Aurora, CO Abstract Acyl-CoA thioesterase 7 (ACOT7) is an intracel- containing unsaturated long (C20)-acyl chains in macro- lular enzyme that converts acyl-CoAs to FFAs. ACOT7 is in- phages, and, although ACOT7 has preferential thioesterase duced by lipopolysaccharide (LPS); thus, we investigated activity toward these lipid species, loss of ACOT7 has no ma- www.jlr.org downstream effects of LPS-induced induction of ACOT7 jor detrimental effect on macrophage inflammatory pheno- and its role in inflammatory settings in myeloid cells. Enzy- types.—Wall, V. Z., S. Barnhart, F. Kramer, J. E. Kanter, matic thioesterase activity assays in WT and ACOT7-deficient A. Vivekanandan-Giri, S. Pennathur, C. Bolego, J. M. Ellis, macrophage lysates indicated that endogenous ACOT7 con- M. -

The Metabolic Serine Hydrolases and Their Functions in Mammalian Physiology and Disease Jonathan Z
REVIEW pubs.acs.org/CR The Metabolic Serine Hydrolases and Their Functions in Mammalian Physiology and Disease Jonathan Z. Long* and Benjamin F. Cravatt* The Skaggs Institute for Chemical Biology and Department of Chemical Physiology, The Scripps Research Institute, 10550 North Torrey Pines Road, La Jolla, California 92037, United States CONTENTS 2.4. Other Phospholipases 6034 1. Introduction 6023 2.4.1. LIPG (Endothelial Lipase) 6034 2. Small-Molecule Hydrolases 6023 2.4.2. PLA1A (Phosphatidylserine-Specific 2.1. Intracellular Neutral Lipases 6023 PLA1) 6035 2.1.1. LIPE (Hormone-Sensitive Lipase) 6024 2.4.3. LIPH and LIPI (Phosphatidic Acid-Specific 2.1.2. PNPLA2 (Adipose Triglyceride Lipase) 6024 PLA1R and β) 6035 2.1.3. MGLL (Monoacylglycerol Lipase) 6025 2.4.4. PLB1 (Phospholipase B) 6035 2.1.4. DAGLA and DAGLB (Diacylglycerol Lipase 2.4.5. DDHD1 and DDHD2 (DDHD Domain R and β) 6026 Containing 1 and 2) 6035 2.1.5. CES3 (Carboxylesterase 3) 6026 2.4.6. ABHD4 (Alpha/Beta Hydrolase Domain 2.1.6. AADACL1 (Arylacetamide Deacetylase-like 1) 6026 Containing 4) 6036 2.1.7. ABHD6 (Alpha/Beta Hydrolase Domain 2.5. Small-Molecule Amidases 6036 Containing 6) 6027 2.5.1. FAAH and FAAH2 (Fatty Acid Amide 2.1.8. ABHD12 (Alpha/Beta Hydrolase Domain Hydrolase and FAAH2) 6036 Containing 12) 6027 2.5.2. AFMID (Arylformamidase) 6037 2.2. Extracellular Neutral Lipases 6027 2.6. Acyl-CoA Hydrolases 6037 2.2.1. PNLIP (Pancreatic Lipase) 6028 2.6.1. FASN (Fatty Acid Synthase) 6037 2.2.2. PNLIPRP1 and PNLIPR2 (Pancreatic 2.6.2. -

Flawed Phospholipid Formation Or Faulty Fatty Acid Oxidation: Determining the Cause of Mitochondrial Dysfunction in Hearts Lacking Acsl1
FLAWED PHOSPHOLIPID FORMATION OR FAULTY FATTY ACID OXIDATION: DETERMINING THE CAUSE OF MITOCHONDRIAL DYSFUNCTION IN HEARTS LACKING ACSL1 Trisha J. Grevengoed A dissertation submitted to the faculty at the University of North Carolina at Chapel Hill in partial fulfillment of the requirements for the degree of Doctor of Philosophy in the Department of Nutrition (Biochemistry) in the School of Public Health. Chapel Hill 2015 Approved by: Rosalind A. Coleman Stephen D. Hursting Liza Makowski Leslie V. Parise Steven H. Zeisel © 2015 Trisha J. Grevengoed ALL RIGHTS RESERVED ii ABSTRACT Trisha J. Grevengoed: Fatty acid activation in cardiac mitochondria: The role of ACSL1 in phospholipid formation and remodeling, substrate switching, and autophagic flux (Under the direction of Rosalind A. Coleman) Cardiovascular disease is the number one cause of death worldwide. In the heart, mitochondria provide up to 95% of energy, with most of this energy coming from metabolism of fatty acids (FA). FA must be converted to acyl-CoAs by acyl-CoA synthetases (ACS) before entry into pathways of β- oxidation or glycerolipid synthesis. ACSL1 contributes more than 90% of total cardiac ACSL activity, and mice with an inducible knockout of ACSL1 (Acsl1T-/-) have impaired cardiac FA oxidation. The effects of loss of ACSL1 on mitochondrial respiratory function, phospholipid formation, or autophagic flux have not yet been studied. Acsl1T-/- hearts contained 3-fold more mitochondria with abnormal structure and displayed lower respiratory function. Because ACSL1 exhibited a strong substrate preference for linoleate (18:2), we investigated the composition of mitochondrial phospholipids. Acsl1T-/- hearts contained 83% less tetralinoleoyl-cardiolipin (CL), the major form present in control hearts. -

A Revised Nomenclature for Mammalian Acyl-Coa Thioesterases/Hydrolases Mary Hunt Dublin Institute of Technology, [email protected]
Dublin Institute of Technology ARROW@DIT Articles School of Biological Sciences 2005-06-01 A revised nomenclature for mammalian acyl-CoA thioesterases/hydrolases Mary Hunt Dublin Institute of Technology, [email protected] Junji Yamada Tokyo University of Pharmacy and Life Science Lois Maltais The Jackson Laboratory Mathew Wright University College London Ernesto Podesta University of Buenos Aires See next page for additional authors Recommended Citation Hunt, M., Maltais, L., Wright, M., Podesta, E., Alexson, S.:A revised nomenclature for mammalian acyl-CoA thioesterases/hydrolases. Journal of Lipid Research, Vol. 45:(10), 2004,pp. 1958-1961. doi:10.1194/jlr.E400002-JLR200 This Article is brought to you for free and open access by the School of Biological Sciences at ARROW@DIT. It has been accepted for inclusion in Articles by an authorized administrator of ARROW@DIT. For more information, please contact [email protected], [email protected]. Authors Mary Hunt, Junji Yamada, Lois Maltais, Mathew Wright, Ernesto Podesta, and Stefan Alexson This article is available at ARROW@DIT: http://arrow.dit.ie/scschbioart/6 1 A revised nomenclature for mammalian acyl-CoA thioesterases/hydrolases. #Mary C. Hunt, ##Junji Yamada, §Lois J. Maltais, *Matthew Wright, §§ Ernesto J.Podesta, and #Stefan E. H. Alexson, #Karolinska Institutet, Department of Laboratory Medicine, Division of Clinical Chemistry C1-74, Karolinska University Hospital at Huddinge, SE- 141 86 Stockholm, Sweden, ##Department of Clinical Biochemistry, Tokyo University of Pharmacy and Life Science, 1432-1 Horinouchi, Hachioji, Tokyo 192-0392, Japan, §The Mouse Genomic Nomenclature Committee (MGNC), Mouse Genome Informatics, The Jackson Laboratory, Bar Harbor, ME, *University College London, London, UK, §§Department of Biochemistry, School of Medicine, University of Buenos Aires, Argentina. -

Monoacylglycerol Lipase Inhibition in Human and Rodent Systems Supports Clinical Evaluation of Endocannabinoid Modulators S
Supplemental material to this article can be found at: http://jpet.aspetjournals.org/content/suppl/2018/10/10/jpet.118.252296.DC1 1521-0103/367/3/494–508$35.00 https://doi.org/10.1124/jpet.118.252296 THE JOURNAL OF PHARMACOLOGY AND EXPERIMENTAL THERAPEUTICS J Pharmacol Exp Ther 367:494–508, December 2018 Copyright ª 2018 The Author(s). This is an open access article distributed under the CC BY Attribution 4.0 International license. Monoacylglycerol Lipase Inhibition in Human and Rodent Systems Supports Clinical Evaluation of Endocannabinoid Modulators s Jason R. Clapper, Cassandra L. Henry, Micah J. Niphakis, Anna M. Knize, Aundrea R. Coppola, Gabriel M. Simon, Nhi Ngo, Rachel A. Herbst, Dylan M. Herbst, Alex W. Reed, Justin S. Cisar, Olivia D. Weber, Andreu Viader, Jessica P. Alexander, Mark L. Cunningham, Todd K. Jones, Iain P. Fraser, Cheryl A. Grice, R. Alan B. Ezekowitz, ’ Gary P. O Neill, and Jacqueline L. Blankman Downloaded from Abide Therapeutics, San Diego, California Received July 26, 2018; accepted October 5, 2018 ABSTRACT Monoacylglycerol lipase (MGLL) is the primary degradative whether selective MGLL inhibition would affect prostanoid pro- jpet.aspetjournals.org enzyme for the endocannabinoid 2-arachidonoylglycerol (2- duction in several human assays known to be sensitive AG). The first MGLL inhibitors have recently entered clinical to cyclooxygenase inhibitors. ABD-1970 robustly elevated brain development for the treatment of neurologic disorders. To 2-AG content and displayed antinociceptive and antipru- support this clinical path, we report the pharmacological ritic activity in a battery of rodent models (ED50 values of characterization of the highly potent and selective MGLL inhibitor 1–2 mg/kg). -

I STRUCTURE and FUNCTION of the PALMITOYLTRANSFERASE
STRUCTURE AND FUNCTION OF THE PALMITOYLTRANSFERASE DHHC20 AND THE ACYL COA HYDROLASE MBLAC2 A Dissertation Presented to the Faculty of the Graduate School Of Cornell University In Partial Fulfillment of the Requirements for the Degree of Doctor of Philosophy By Martin Ian Paguio Malgapo December 2019 i © 2019 Martin Ian Paguio Malgapo ii STRUCTURE AND FUNCTION OF THE PALMITOYLTRANSFERASE DHHC20 AND THE ACYL COA HYDROLASE MBLAC2 Martin Ian Paguio Malgapo, Ph.D. Cornell University 2019 My graduate research has focused on the enzymology of protein S-palmitoylation, a reversible posttranslational modification catalyzed by DHHC palmitoyltransferases. When I started my thesis work, the structure of DHHC proteins was not known. I sought to purify and crystallize a DHHC protein, identifying DHHC20 as the best target. While working on this project, I came across a protein of unknown function called metallo-β-lactamase domain-containing protein 2 (MBLAC2). A proteomic screen utilizing affinity capture mass spectrometry suggested an interaction between MBLAC2 (bait) and DHHC20 (hit) in HEK-293 cells. This finding interested me initially from the perspective of finding an interactor that could help stabilize DHHC20 into forming better quality crystals as well as discovering a novel protein substrate for DHHC20. I was intrigued by MBLAC2 upon learning that this protein is predicted to be palmitoylated by multiple proteomic screens. Additionally, sequence analysis predicts MBLAC2 to have thioesterase activity. Taken together, studying a potential new thioesterase that is itself palmitoylated was deemed to be a worthwhile project. When the structure of DHHC20 was published in 2017, I decided to switch my efforts to characterizing MBLAC2. -

Structural Basis for Recruitment of Tandem Hotdog Domains in Acyl-Coa Thioesterase 7 and Its Role in Inflammation
Structural basis for recruitment of tandem hotdog domains in acyl-CoA thioesterase 7 and its role in inflammation Jade K. Forwood*†‡, Anil S. Thakur*, Gregor Guncar*§¶, Mary Marfori*, Dmitri Mouradov*, Weining Meng*§, Jodie Robinson§ʈ, Thomas Huber*, Stuart Kellie*§ʈ, Jennifer L. Martin*§**, David A. Hume*§ʈ**††, and Bostjan Kobe*‡§** *School of Molecular and Microbial Sciences, §Institute for Molecular Bioscience, ʈCooperative Research Centre for Chronic Inflammatory Diseases, and **Australian Research Council Special Research Centre for Functional and Applied Genomics, University of Queensland, Brisbane, Queensland 4072, Australia Edited by Gregory A. Petsko, Brandeis University, Waltham, MA, and approved May 10, 2007 (received for review February 2, 2007) Acyl-CoA thioesterases (Acots) catalyze the hydrolysis of fatty long-chain acyl-CoA substrates with fatty acid chains of 8–16 acyl-CoA to free fatty acid and CoA and thereby regulate lipid carbon atoms (C8–C16) (7, 13). Acot7 contains a pair of fused metabolism and cellular signaling. We present a comprehensive thioesterase domains that share Ϸ30% sequence identity, with each structural and functional characterization of mouse acyl-CoA thio- thioesterase domain predicted to have the hotdog fold structure esterase 7 (Acot7). Whereas prokaryotic homologues possess a with an ␣-helix sausage wrapped by a -sheet bun (14–16). single thioesterase domain, mammalian Acot7 contains a pair of Here, we present the crystal structures of the N- and C- domains in tandem. We determined the crystal structures of both terminal thioesterase domains (N- and C-domains) of mouse the N- and C-terminal domains of the mouse enzyme, and inferred Acot7, refined at 1.8 and 2.4 Å resolution, respectively. -

The Emerging Role of Acyl-Coa Thioesterases and Acyltransferases in Regulating Peroxisomal Lipid Metabolism☆
View metadata, citation and similar papers at core.ac.uk brought to you by CORE provided by Elsevier - Publisher Connector Biochimica et Biophysica Acta 1822 (2012) 1397–1410 Contents lists available at SciVerse ScienceDirect Biochimica et Biophysica Acta journal homepage: www.elsevier.com/locate/bbadis Review The emerging role of acyl-CoA thioesterases and acyltransferases in regulating peroxisomal lipid metabolism☆ Mary C. Hunt a,⁎, Marina I. Siponen b,1, Stefan E.H. Alexson c a Dublin Institute of Technology, School of Biological Sciences, College of Sciences & Health, Kevin Street, Dublin 8, Ireland b Department of Medical Biochemistry and Biophysics, Structural Genomics Consortium, Karolinska Institutet, SE-171 77 Stockholm, Sweden c Karolinska Institutet, Department of Laboratory Medicine, Division of Clinical Chemistry, Karolinska University Hospital at Huddinge, SE-141 86 Stockholm, Sweden article info abstract Article history: The importance of peroxisomes in lipid metabolism is now well established and peroxisomes contain approx- Received 15 November 2011 imately 60 enzymes involved in these lipid metabolic pathways. Several acyl-CoA thioesterase enzymes Received in revised form 3 March 2012 (ACOTs) have been identified in peroxisomes that catalyze the hydrolysis of acyl-CoAs (short-, medium-, Accepted 16 March 2012 long- and very long-chain), bile acid-CoAs, and methyl branched-CoAs, to the free fatty acid and coenzyme Available online 23 March 2012 A. A number of acyltransferase enzymes, which are structurally and functionally related to ACOTs, have also been identified in peroxisomes, which conjugate (or amidate) bile acid-CoAs and acyl-CoAs to amino acids, Keywords: Acyl-CoA thioesterase resulting in the production of amidated bile acids and fatty acids. -

Comparative Analysis of Cerebrospinal Fluid Metabolites in Alzheimer's
Nagata et al. Biomarker Research (2018) 6:5 DOI 10.1186/s40364-018-0119-x RESEARCH Open Access Comparative analysis of cerebrospinal fluid metabolites in Alzheimer’s disease and idiopathic normal pressure hydrocephalus in a Japanese cohort Yuki Nagata1* , Akiyoshi Hirayama2, Satsuki Ikeda2, Aoi Shirahata2, Futaba Shoji2, Midori Maruyama2, Mitsunori Kayano3, Masahiko Bundo4, Kotaro Hattori5, Sumiko Yoshida5, Yu-ichi Goto5, Katsuya Urakami6, Tomoyoshi Soga2, Kouichi Ozaki1 and Shumpei Niida1 Abstract Background: Alzheimer’s disease (AD) is a most common dementia in elderly people. Since AD symptoms resemble those of other neurodegenerative diseases, including idiopathic normal pressure hydrocephalus (iNPH), it is difficult to distinguish AD from iNPH for a precise and early diagnosis. iNPH is caused by the accumulation of cerebrospinal fluid (CSF) and involves gait disturbance, urinary incontinence, and dementia. iNPH is treatable with shunt operation which removes accumulated CSF from the brain ventricles. Methods: We performed metabolomic analysis in the CSF of patients with AD and iNPH with capillary electrophoresis-mass spectrometry. We assessed metabolites to discriminate between AD and iNPH with Welch’s t-test, receiver operating characteristic (ROC) curve analysis, and multiple logistic regression analysis. Results: We found significant increased levels of glycerate and N-acetylneuraminate and significant decreased levels of serine and 2-hydroxybutyrate in the CSF of patients with AD compared to the CSF of patients with iNPH. The ROC curve analysis with these four metabolites showed that the area under the ROC curve was 0.90, indicating good discrimination between AD and iNPH. Conclusions: This study identified four metabolites that could possibly discriminate between AD and iNPH, whichpreviousresearchhasshownarecloselyrelatedto the risk factors, pathogenesis, and symptoms of AD. -
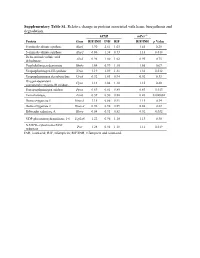
Supplementary Table S1. Relative Change in Proteins Associated with Heme Biosynthesis and Degradation
Supplementary Table S1. Relative change in proteins associated with heme biosynthesis and degradation. hPXR mPxr–/– Protein Gene RIF/INH INH RIF RIF/INH p Value 5-aminolevulinate synthase Alas1 1.90 2.61 1.05 1.41 0.28 5-aminolevulinate synthase Alas2 0.86 1.38 0.73 1.18 0.018 Delta-aminolevulinic acid Alad 0.96 1.00 1.02 0.95 0.75 dehydratase Porphobilinogen deaminase Hmbs 1.04 0.99 1.10 1.05 0.67 Uroporphyrinogen-III synthase Uros 1.19 1.09 1.31 1.38 0.012 Uroporphyrinogen decarboxylase Urod 0.92 1.03 0.94 0.92 0.33 Oxygen-dependent Cpox 1.13 1.04 1.18 1.15 0.20 coproporphyrinogen-III oxidase, Protoporphyrinogen oxidase Ppox 0.69 0.81 0.85 0.83 0.013 Ferrochelatase, Fech 0.39 0.50 0.88 0.43 0.000002 Heme oxygenase 1 Hmox1 1.15 0.86 0.91 1.11 0.34 Heme oxygenase 2 Hmox2 0.96 0.98 0.89 0.88 0.22 Biliverdin reductase A Blvra 0.84 0.92 0.82 0.92 0.032 UDP-glucuronosyltransferase 1-6 Ugt1a6 1.22 0.96 1.10 1.13 0.30 NADPH--cytochrome P450 Por 1.28 0.92 1.18 1.12 0.019 reductase INH, isoniazid; RIF, rifampicin; RIF/INH, rifampicin and isoniazid. Supplementary Table S2. Relative change in protein nuclear receptors. hPXR mPxr–/– Protein Gene RIF/INH INH RIF RIF/INH p Value Aryl hydrocarbon receptor Ahr 1.09 0.91 1.00 1.26 0.092 Hepatocyte nuclear factor Hnf1a 0.87 0.97 0.82 0.79 0.027 1-alpha Hepatocyte nuclear factor Hnf4a 0.95 1.05 0.97 1.08 0.20 4-alpha Oxysterols receptor LXR- Nr1h3 0.94 1.16 1.03 1.02 0.42 alpha Bile acid receptor Nr1h4 1.05 1.17 0.98 1.19 0.12 Retinoic acid receptor Rxra 0.88 1.03 0.83 0.95 0.12 RXR-alpha Peroxisome proliferator- -

Differential Regulation of Gene Expression by Cholesterol
Supplemental material to this article can be found at: http://jpet.aspetjournals.org/content/suppl/2016/05/25/jpet.116.233312.DC1 1521-0103/358/2/216–229$25.00 http://dx.doi.org/10.1124/jpet.116.233312 THE JOURNAL OF PHARMACOLOGY AND EXPERIMENTAL THERAPEUTICS J Pharmacol Exp Ther 358:216–229, August 2016 Copyright ª 2016 by The American Society for Pharmacology and Experimental Therapeutics Differential Regulation of Gene Expression by Cholesterol Biosynthesis Inhibitors That Reduce (Pravastatin) or Enhance (Squalestatin 1) Nonsterol Isoprenoid Levels in Primary Cultured Mouse and Rat Hepatocytes s Elizabeth A. Rondini, Zofia Duniec-Dmuchowski, Daniela Cukovic, Alan A. Dombkowski, and Thomas A. Kocarek Institute of Environmental Health Sciences (E.A.R., Z.D.-D., T.A.K.), and Department of Pediatrics, Division of Clinical Pharmacology and Toxicology (D.C., A.A.D.), Wayne State University, Detroit, Michigan Downloaded from Received March 30, 2016; accepted May 24, 2016 ABSTRACT Squalene synthase inhibitors (SSIs), such as squalestatin 1 (SQ1), directly, 162 orthologs were found to be differentially coregu- reduce cholesterol biosynthesis but cause the accumulation of lated between the two treatments. Genes involved in cholesterol jpet.aspetjournals.org isoprenoids derived from farnesyl pyrophosphate (FPP), which and unsaturated fatty acid biosynthesis were up-regulated by can modulate the activity of nuclear receptors, including the both inhibitors, consistent with cholesterol depletion; however, constitutive androstane receptor (CAR), farnesoid X receptor, the extent of induction was greater in rat than in mouse hepa- and peroxisome proliferator-activated receptors (PPARs). In tocytes. SQ1 induced several orthologs associated with micro- comparison, 3-hydroxy-3-methylglutaryl-coenzyme A reduc- somal, peroxisomal, and mitochondrial fatty acid oxidation and tase inhibitors (e.g., pravastatin) inhibit production of both repressed orthologs involved in cell cycle regulation. -

MOL #82305 TITLE PAGE Title: Induced CYP3A4 Expression In
Downloaded from molpharm.aspetjournals.org at ASPET Journals on September 28, 2021 1 This article has not been copyedited and formatted. The final version may differ from this version. This article has not been copyedited and formatted. The final version may differ from this version. This article has not been copyedited and formatted. The final version may differ from this version. This article has not been copyedited and formatted. The final version may differ from this version. This article has not been copyedited and formatted. The final version may differ from this version. This article has not been copyedited and formatted. The final version may differ from this version. This article has not been copyedited and formatted. The final version may differ from this version. This article has not been copyedited and formatted. The final version may differ from this version. This article has not been copyedited and formatted. The final version may differ from this version. This article has not been copyedited and formatted. The final version may differ from this version. This article has not been copyedited and formatted. The final version may differ from this version. This article has not been copyedited and formatted. The final version may differ from this version. This article has not been copyedited and formatted. The final version may differ from this version. This article has not been copyedited and formatted. The final version may differ from this version. This article has not been copyedited and formatted. The final version may differ from this version. This article has not been copyedited and formatted.