Structure and Function of A.Nidulans Psi Factor Producing Oxygenase A
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Korrdel1lic Avhandl Ahmad Zayny
Faculty of Pharmacy, Uppsala University LICENTIATE THESES 50 Vitamin D metabolism in osteoblast-like cells: effects of drugs on inactivation by CYP24A1 by Ahmad Zayny 2018 Faculty of Pharmacy, Uppsala University LICENTIATE THESES 50 Summary Vitamin D metabolism in osteoblast-like cells: effects of drugs on inactivation by CYP24A1 by Ahmad Zayny 2018 2 This licentiate thesis will be defended on Wednesday, May 30, 2018 at 13:15 in C4:305, BMC, Husargatan 3, Uppsala. The presentation and discussion will be conducted in Swedish. Abstract Zayny, A. 2018 Vitamin D metabolism in osteoblast-like cells: effects of drugs on inactivation by CYP24A1 Vitamin D is essential for bone function, and deficiency in active vitamin D hormone can lead to bone disorders. Long-term treatment with glucocorticoids and antiretroviral drugs used to treat HIV infection, results in osteoporosis and increased risk of fractures. Much remains unclear regarding the effects of these compounds in bone cells. In the current study, human osteosarcoma Saos-2 cells and primary human osteoblasts were found to express mRNA for the vitamin D receptor as well as activating and deactivating enzymes in vitamin D3 metabolism. These bone cells exhibited CYP24A1-mediated 24-hydroxylation, involved in deactivation of the active vitamin form. However, bioactivating vitamin D3 hydroxylase activities were not detected in either of these cells, indicating that local vitamin D bioactivation is not significant in osteoblasts. Several glucocorticoids and antiretroviral drugs, including prednisolone, efavirenz and ritonavir, down regulated CYP24A1 mRNA expression. Prednisolone and ritonavir also down regulated CYP24A1-mediated 24-hydroxylase activity in both Saos-2 and primary human osteoblasts. -

And Dioxygenase Chemistries in a Single Active Site Promotes Heme Degradation
Unique coupling of mono- and dioxygenase chemistries in a single active site promotes heme degradation Toshitaka Matsuia,1, Shusuke Nambua, Celia W. Gouldingb,c, Satoshi Takahashia, Hiroshi Fujiid, and Masao Ikeda-Saitoa,1 aInstitute of Multidisciplinary Research for Advanced Materials, Tohoku University, Katahira, Aoba, Sendai 980-8577, Japan; bDepartment of Molecular Biology and Biochemistry, University of California, Irvine, CA 92697; cDepartment of Pharmaceutical Sciences, University of California, Irvine, CA 92697; and dDepartment of Chemistry, Graduate School of Humanities and Sciences, Nara Women’s University, Kitauoyanishi, Nara 630-8506, Japan Edited by Harry B. Gray, California Institute of Technology, Pasadena, CA, and approved February 26, 2016 (received for review November 26, 2015) − + Bacterial pathogens must acquire host iron for survival and colo- RH + O2 + 2e + 2H → ROH + H2O, [1] nization. Because free iron is restricted in the host, numerous pathogens have evolved to overcome this limitation by using a R + O → RðOÞ . [2] family of monooxygenases that mediate the oxidative cleavage of 2 2 heme into biliverdin, carbon monoxide, and iron. However, the Biological heme degradation proceeds through a unique self- etiological agent of tuberculosis, Mycobacterium tuberculosis, ac- oxidation mechanism where the substrate heme activates O complishes this task without generating carbon monoxide, which 2 potentially induces its latent state. Here we show that this unusual molecules. The canonical enzyme, heme oxygenase (HO), de- heme degradation reaction proceeds through sequential mono- grades heme into ferrous iron, carbon monoxide (CO), and bil- and dioxygenation events within the single active center of MhuD, iverdin by three successive monooxygenation reactions (Fig. 1A) a mechanism unparalleled in enzyme catalysis. -

Springer Handbook of Enzymes
Dietmar Schomburg and Ida Schomburg (Eds.) Springer Handbook of Enzymes Volume 25 Class 1 • Oxidoreductases X EC 1.9-1.13 co edited by Antje Chang Second Edition 4y Springer Index of Recommended Enzyme Names EC-No. Recommended Name Page 1.13.11.50 acetylacetone-cleaving enzyme 673 1.10.3.4 o-aminophenol oxidase 149 1.13.12.12 apo-/?-carotenoid-14',13'-dioxygenase 732 1.13.11.34 arachidonate 5-lipoxygenase 591 1.13.11.40 arachidonate 8-lipoxygenase 627 1.13.11.31 arachidonate 12-lipoxygenase 568 1.13.11.33 arachidonate 15-lipoxygenase 585 1.13.12.1 arginine 2-monooxygenase 675 1.13.11.13 ascorbate 2,3-dioxygenase 491 1.10.2.1 L-ascorbate-cytochrome-b5 reductase 79 1.10.3.3 L-ascorbate oxidase 134 1.11.1.11 L-ascorbate peroxidase 257 1.13.99.2 benzoate 1,2-dioxygenase (transferred to EC 1.14.12.10) 740 1.13.11.39 biphenyl-2,3-diol 1,2-dioxygenase 618 1.13.11.22 caffeate 3,4-dioxygenase 531 1.13.11.16 3-carboxyethylcatechol 2,3-dioxygenase 505 1.13.11.21 p-carotene 15,15'-dioxygenase (transferred to EC 1.14.99.36) 530 1.11.1.6 catalase 194 1.13.11.1 catechol 1,2-dioxygenase 382 1.13.11.2 catechol 2,3-dioxygenase 395 1.10.3.1 catechol oxidase 105 1.13.11.36 chloridazon-catechol dioxygenase 607 1.11.1.10 chloride peroxidase 245 1.13.11.49 chlorite O2-lyase 670 1.13.99.4 4-chlorophenylacetate 3,4-dioxygenase (transferred to EC 1.14.12.9) . -

Unique Coupling of Mono- and Dioxygenase Chemistries in a Single Active Site Promotes Heme Degradation
Unique coupling of mono- and dioxygenase chemistries in a single active site promotes heme degradation Toshitaka Matsuia,1, Shusuke Nambua, Celia W. Gouldingb,c, Satoshi Takahashia, Hiroshi Fujiid, and Masao Ikeda-Saitoa,1 aInstitute of Multidisciplinary Research for Advanced Materials, Tohoku University, Katahira, Aoba, Sendai 980-8577, Japan; bDepartment of Molecular Biology and Biochemistry, University of California, Irvine, CA 92697; cDepartment of Pharmaceutical Sciences, University of California, Irvine, CA 92697; and dDepartment of Chemistry, Graduate School of Humanities and Sciences, Nara Women’s University, Kitauoyanishi, Nara 630-8506, Japan Edited by Harry B. Gray, California Institute of Technology, Pasadena, CA, and approved February 26, 2016 (received for review November 26, 2015) − + Bacterial pathogens must acquire host iron for survival and colo- RH + O2 + 2e + 2H → ROH + H2O, [1] nization. Because free iron is restricted in the host, numerous pathogens have evolved to overcome this limitation by using a R + O → RðOÞ . [2] family of monooxygenases that mediate the oxidative cleavage of 2 2 heme into biliverdin, carbon monoxide, and iron. However, the Biological heme degradation proceeds through a unique self- etiological agent of tuberculosis, Mycobacterium tuberculosis, ac- oxidation mechanism where the substrate heme activates O complishes this task without generating carbon monoxide, which 2 potentially induces its latent state. Here we show that this unusual molecules. The canonical enzyme, heme oxygenase (HO), de- heme degradation reaction proceeds through sequential mono- grades heme into ferrous iron, carbon monoxide (CO), and bil- and dioxygenation events within the single active center of MhuD, iverdin by three successive monooxygenation reactions (Fig. 1A) a mechanism unparalleled in enzyme catalysis. -

Discovery of Novel Fatty Acid Dioxygenases and Cytochromes P450
Digital Comprehensive Summaries of Uppsala Dissertations from the Faculty of Pharmacy 176 Discovery of Novel Fatty Acid Dioxygenases and Cytochromes P450 Mechanisms of Oxylipin Biosynthesis in Pathogenic Fungi INGA HOFFMANN ACTA UNIVERSITATIS UPSALIENSIS ISSN 1651-6192 ISBN 978-91-554-8739-3 UPPSALA urn:nbn:se:uu:diva-206199 2013 Dissertation presented at Uppsala University to be publicly examined in B21, BMC, Husargatan 3, Uppsala, Friday, October 18, 2013 at 09:15 for the degree of Doctor of Philosophy (Faculty of Pharmacy). The examination will be conducted in English. Abstract Hoffmann, I. 2013. Discovery of Novel Fatty Acid Dioxygenases and Cytochromes P450: Mechanisms of Oxylipin Biosynthesis in Pathogenic Fungi. Acta Universitatis Upsaliensis. Digital Comprehensive Summaries of Uppsala Dissertations from the Faculty of Pharmacy 176. 67 pp. Uppsala. ISBN 978-91-554-8739-3. Dioxygenase-cytochrome P450 (DOX-CYP) fusion enzymes are present in diverse human and plant pathogenic fungi. They oxygenate fatty acids to lipid mediators which have regulatory functions in fungal development and toxin production. These enzymes catalyze the formation of fatty acid hydroperoxides which are subsequently converted by the P450 activities or reduced to the corresponding alcohols. The N-terminal DOX domains show catalytic and structural homology to mammalian cyclooxygenases, which belong to the most thoroughly studied human enzymes. 7,8-Linoleate diol synthase (LDS) of the plant pathogenic fungus Gaeumannomyces graminis was the first characterized member of the DOX-CYP fusion enzyme family. It catalyzes the conversion of linoleic acid to 8R-hydroperoxylinoleic acid (HPODE) and subsequently to 7S,8S- dihydroxylinoleic acid by its DOX and P450 domains, respectively. -

Untersuchung Der Stereospezifität Und Positionsspezifität Von Wirbeltier-Lipoxygenasen
Aus dem Institut für Biochemie der Medizinischen Fakultät Charité – Universitätsmedizin Berlin DISSERTATION Untersuchung der Stereospezifität und Positionsspezifität von Wirbeltier-Lipoxygenasen zur Erlangung des akademischen Grades Doctor medicinae (Dr. med.) vorgelegt der Medizinischen Fakultät Charité – Universitätsmedizin Berlin von Christian Andreas Jansen aus Essen Datum der Promotion: 14.02.2014 INHALTSVERZEICHNIS ABKÜRZUNGEN ZUSAMMENFASSUNG ABSTRACT 1. EINLEITUNG . 1 1.1. Vorkommen und Klassifizierung von Lipoxygenasen . 1 1.1.1. Funktionsweise von LOXen . 2 1.1.2. Verbreitung von LOXen und LOX-Nomenklatur . 4 1.1.2.1. Verbreitung von LOXen . 4 1.1.2.2. LOX-Nomenklatur . 6 1.1.3. Biologische Bedeutung verschiedener LOX-Isoformen . 7 1.1.3.1. LOXen bei der Entzündungsreaktion . 10 1.1.3.2. LOXen bei Atherosklerose . 10 1.1.3.3. LOXen beim Asthma bronchiale . 13 1.1.3.4. LOXen bei der Kanzerogenese . 14 1.1.3.5. LOXen bei der epidermalen Differenzierung . 15 1.1.3.6. Sonstige Funktionen von LOXen . 16 1.2. Struktur-Funktionsbeziehung bei Lipoxygenasen . 17 1.2.1. Strukturelle Basis der LOX-Katalyse . 18 1.2.2. Strukturelle Grundlage der Reaktionsspezifität von LOXen . 18 1.2.2.1. Determinanten der Positionsspezifität . 18 1.2.2.2. Determinanten der Stereospezifität . 21 1.2.3. Kristall- und Lösungsstruktur der ALOX15 des Kaninchens . 24 1.2.4. Kristallstruktur der ALOX15 des Schweins . 26 1.2.5. Kristallstruktur der humanen ALOX5 . 28 1.2.6. Kristallstruktur der 11R-LOX der Weichkoralle Gersemia fruticosa . 30 1.3. Ziele der Arbeit . 32 2. MATERIALEN UND METHODEN . 33 2.1. Materialen . 33 2.1.1. Chemikalien und Biochemikalien . -

Linoleate Diol Synthase Related Enzymes of the Human Pathogens Histoplasma Capsulatum and Blastomyces Dermatitidis
Archives of Biochemistry and Biophysics 696 (2020) 108669 Contents lists available at ScienceDirect Archives of Biochemistry and Biophysics journal homepage: www.elsevier.com/locate/yabbi Linoleate diol synthase related enzymes of the human pathogens Histoplasma capsulatum and Blastomyces dermatitidis Ernst H. Oliw Division of Biochemical Pharmacology, Department of Pharmaceutical Biosciences, Uppsala University, Box 591, SE 751 24, Uppsala, Sweden ARTICLE INFO ABSTRACT Keywords: Histoplasma capsulatum is an ascomyceteous fungus and a human lung pathogen, which is present in river valleys Cytochrome P450 of the Americas and other continents. H. capsulatum and two related human pathogens, Blasmomyces dermatitidis Linoleate diol synthase and Paracoccidioides brasiliensis, belongs to the Ajellomycetaceae family. The genomes of all three species code Lipid metabolism for three homologous and tentative enzymes of the linoleate diol synthase (LDS) family of fusion enzymes with Mass spectrometry dioxygenase (DOX) and cytochrome P450 domains. One group aligned closely with 8R-DOX-5,8-LDS of Asper Oxygenation mechanism Paracoccidioides brasiliensis gilli, which oxidizes linoleic acid to 5S,8R-dihydroxylinoleic acid; this group was not further investigated. The second group aligned with 10R-DOX-epoxy alcohol synthase (EAS) of plant pathogens. Expression of this enzyme from B. dermatitidis revealed only 10R-DOX activities, i.e., oxidation of linoleic acid to 10R-hydroperoxy-8E,12Z- octadecadienoic acid. The third group aligned in a separate entity. Expression of these enzymes of H. capsulatum and B. dermatitidis revealed no DOX activities, but both enzymes transformed 13S-hydroperoxy-9Z,11E-octade cadienoic acid efficiently to 12(13S)epoxy-11-hydroperoxy-9Z-octadecenoic acid. Other 13-hydroperoxides of linoleic and α-linolenic acids were transformed with less efficiency and the 9-hydroperoxides of linoleic acid were not transformed. -
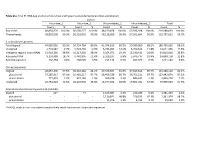
Table S1a. Trial #1 RNA-Seq Analysis of Virus-Free and Hypovirus
Table S1a. Trial #1 RNA-Seq analysis of virus-free and hypovirus-transfected Sclerotinia sclerotiorum Library Virus-free_1 Virus-free_2 Virus-infected_1 Virus-infected_2 Total Count % Count % Count % Count % Count % Raw reads 48,854,170 100.0% 56,568,277 100.0% 48,169,458 100.0% 57,396,928 100.0% 210,988,833 100.0% Filtered reads 48,810,008 99.9% 56,518,010 99.9% 48,118,009 99.9% 57,341,604 99.9% 210,787,631 99.9% S. sclerotiorum genome Total aligned 44,085,026 90.3% 50,724,764 89.8% 41,374,916 86.0% 50,580,960 88.2% 186,765,666 88.6% Unaligned 4,724,982 9.7% 5,793,246 10.3% 6,743,093 14.0% 6,760,644 11.8% 24,021,965 11.5% Intergenic regions (non-rRNA) 13,918,285 28.5% 16,317,039 28.9% 9,354,071 19.4% 15,230,421 26.6% 54,819,816 25.8% Ribosomal RNA 9,150,409 18.7% 6,442,905 11.4% 3,329,107 6.8% 5,976,737 10.4% 24,899,158 11.8% Retrotransposons 202,264 0.4% 268,505 0.5% 251,134 0.5% 499,377 0.9% 1,221,280 0.6% Coding sequences Aligned 28,057,390 57.5% 36,310,362 64.2% 29,370,507 61.0% 37,649,843 65.7% 131,388,102 62.1% plus strand 27,285,917 97.3% 35,481,217 97.7% 28,409,529 96.7% 36,766,211 97.7% 127,942,874 97.3% minus strand 771,473 2.7% 829,145 2.3% 960,978 3.3% 883,632 2.3% 3,445,228 2.7% Unaligned 20,752,618 42.5% 20,207,648 35.8% 18,747,502 39.0% 19,691,761 34.3% 79,399,529 37.9% Sclerotinia sclerotiorum hypovirus 2L (SsHV2L) Aligned 62* - 77 - 1,165,087 2.4% 216,286 0.4% 1,381,450 1.4% plus strand - - - - 1,150,841 98.8% 210,533 97.3% 1,361,374 98.1% minus strand - - - - 14,246 1.2% 5,753 2.7% 19,999 1.9% *SsHV2L reads in non-inoculated samples likely result from errors in barcode sequences. -

A New Class of Fatty Acid Allene Oxide Formed by the DOX-P450 Fusion Proteins of Human and Plant Pathogenic Fungi, C
Supplemental Material can be found at: http://www.jlr.org/content/suppl/2016/06/09/jlr.M068981.DC1 .html A new class of fatty acid allene oxide formed by the DOX-P450 fusion proteins of human and plant pathogenic fungi, C. immitis and Z. tritici Ernst H. Oliw,1 Marc Aragó, Yang Chen, and Fredrik Jernerén Division of Biochemical Pharmacology, Department of Pharmaceutical Biosciences, Uppsala University, SE-751 24 Uppsala, Sweden Abstract Linoleate dioxygenase-cytochrome P450 (DOX- Eicosanoids in humans and oxylipins in plants and fungi Downloaded from CYP) fusion enzymes are common in pathogenic fungi. The designate oxygenated unsaturated C20 and C18 fatty acids DOX domains form hydroperoxy metabolites of 18:2n-6, and many of them exert potent biological actions (1–3). which can be transformed by the CYP domains to 1,2- or Fungal oxylipins can be formed by dioxygenation of C 1,4-diols, epoxy alcohols, or to allene oxides. We have char- 18 fatty acids by two groups of enzymes, lipoxygenases (LOXs) acterized two novel allene oxide synthases (AOSs), namely, recombinant 8R-DOX-AOS of Coccidioides immitis (causing and heme-containing dioxygenases (DOXs) (2). The LOXs www.jlr.org valley fever) and 8S-DOX-AOS of Zymoseptoria tritici (causing contain catalytic Fe or Mn, and oxidize unsaturated fatty septoria tritici blotch of wheat). The 8R-DOX-AOS oxidized acids to hydroperoxides by hydrogen abstraction at bis- 18:2n-6 sequentially to 8R-hydroperoxy-9Z,12Z-octadeca- allylic positions (4–8). The heme-containing DOXs belong dienoic acid (8R-HPODE) and to an allene oxide, 8R(9)- to the cyclooxygenase (COX) gene family (9). -

Untersuchungen Zum Funktionellen Einfluss Von Einzelnukleotidvariationen in Humanen Lipoxygenasen Und Anderen Genen Des LOX-Stoffwechsels
Untersuchungen zum funktionellen Einfluss von Einzelnukleotidvariationen in humanen Lipoxygenasen und anderen Genen des LOX-Stoffwechsels DISSERTATION zur Erlangung des akademischen Grades des Doktors der Naturwissenschaften (Dr. rer. nat.) eingereicht im Fachbereich Biologie, Chemie, Pharmazie der Freien Universität Berlin vorgelegt von Diplom-Biochemiker Thomas Horn aus Berlin 2014 Diese Arbeit wurde zwischen dem 01.01.2011 und 31.12.2013 am Institut für Biochemie der Medizinischen Fakultät der Charité – Universitätsmedizin Berlin unter der Leitung von Prof. Dr. sc. med. Hartmut Kühn angefertigt. 1. Gutachter: Prof. Dr. Rupert Mutzel 2. Gutachter: Prof. Dr. Hartmut Kühn Disputation am 18.03.2014 Danksagung Mein besonderer Dank gilt Herrn Prof. Dr. Hartmut Kühn für die freundliche Überlassung des Arbeitsthemas. Außerdem möchte ich mich bei ihm für die gute wissenschaftliche Betreuung und die erfolgreiche Zusammenarbeit bedanken. Er war stets offen für Diskussionen, Fragen und Probleme, was für das erfolgreiche Gelingen dieser Arbeit entscheidend war. Ich danke Herrn Dr. Igor Ivanov und Frau Dr. Almerinda Di Venere für die Durchführung der CD- und Fluoreszenzmessungen. Insbesondere möchte ich mich bei Igor für seine stetige Unterstützung bedanken. Er hat mir alle wichtigen Dinge über die experimentelle Herstellung, Reinigung und Charakterisierung von Lipoxygenasen beigebracht. Danke auch für das Lesen der Manuskripte sowie der Arbeit. Daneben bedanke ich mich recht herzlich bei Frau Dr. Astrid Borchert für die stets interessanten wissenschaftlichen Gespräche, die schönen Mittagspausen und ihre immer fortwährende moralische Unterstützung. Ich bedanke mich auch bei Frau Dr. Dagmar Heydeck und Herrn Dr. Christoph Ufer für die stets gute wissenschaftliche Beratung und ihre Hilfe bei den wissenschaftlichen Arbeiten. Ich danke Frau Sabine Stehling und Frau Monika Anton für die hervorragende technische Unterstützung und ihre viele Geduld mit mir. -

In Silico/In Vivo Insights Into the Functional and Evolutionary Pathway of Pseudomonas Aeruginosa Oleate-Diol Synthase
RESEARCH ARTICLE In Silico/In Vivo Insights into the Functional and Evolutionary Pathway of Pseudomonas aeruginosa Oleate-Diol Synthase. Discovery of a New Bacterial Di-Heme Cytochrome C Peroxidase Subfamily Mónica Estupiñán1,2, Daniel Álvarez-García3,4, Xavier Barril3,4,5, Pilar Diaz2*, Angeles Manresa1 1 Unitat de Microbiologia i Parasitologia Sanitàries, Facultat de Farmàcia, University of Barcelona, Barcelona, Spain, 2 Department of Microbiology, Faculty of Biology, University of Barcelona, Barcelona, Spain, 3 Departament de Fisicoquímica, Facultat de Farmàcia, Universitat de Barcelona, Barcelona, Spain, 4 Institut de Biomedicina de la Universitat de Barcelona (IBUB), PCB-Edifici Hèlix Baldiri Reixac, Barcelona, Spain, 5 Catalan Institution for Research and Advanced Studies (ICREA), Passeig Lluís Companys, Barcelona, Spain OPEN ACCESS * [email protected] Citation: Estupiñán M, Álvarez-García D, Barril X, Diaz P, Manresa A (2015) In Silico/In Vivo Insights into the Functional and Evolutionary Pathway of Pseudomonas aeruginosa Oleate-Diol Synthase. Abstract Discovery of a New Bacterial Di-Heme Cytochrome C Peroxidase Subfamily. PLoS ONE 10(7): e0131462. As previously reported, P. aeruginosa genes PA2077 and PA2078 code for 10S-DOX (10S- doi:10.1371/journal.pone.0131462 Dioxygenase) and 7,10-DS (7,10-Diol Synthase) enzymes involved in long-chain fatty acid Editor: Ligia O Martins, Universidade Nova de oxygenation through the recently described oleate-diol synthase pathway. Analysis of the Lisboa, PORTUGAL amino acid sequence of both enzymes revealed the presence of two heme-binding motifs Received: January 20, 2015 (CXXCH) on each protein. Phylogenetic analysis showed the relation of both proteins to Accepted: June 2, 2015 bacterial di-heme cytochrome c peroxidases (Ccps), similar to Xanthomonas sp. -

All Enzymes in BRENDA™ the Comprehensive Enzyme Information System
All enzymes in BRENDA™ The Comprehensive Enzyme Information System http://www.brenda-enzymes.org/index.php4?page=information/all_enzymes.php4 1.1.1.1 alcohol dehydrogenase 1.1.1.B1 D-arabitol-phosphate dehydrogenase 1.1.1.2 alcohol dehydrogenase (NADP+) 1.1.1.B3 (S)-specific secondary alcohol dehydrogenase 1.1.1.3 homoserine dehydrogenase 1.1.1.B4 (R)-specific secondary alcohol dehydrogenase 1.1.1.4 (R,R)-butanediol dehydrogenase 1.1.1.5 acetoin dehydrogenase 1.1.1.B5 NADP-retinol dehydrogenase 1.1.1.6 glycerol dehydrogenase 1.1.1.7 propanediol-phosphate dehydrogenase 1.1.1.8 glycerol-3-phosphate dehydrogenase (NAD+) 1.1.1.9 D-xylulose reductase 1.1.1.10 L-xylulose reductase 1.1.1.11 D-arabinitol 4-dehydrogenase 1.1.1.12 L-arabinitol 4-dehydrogenase 1.1.1.13 L-arabinitol 2-dehydrogenase 1.1.1.14 L-iditol 2-dehydrogenase 1.1.1.15 D-iditol 2-dehydrogenase 1.1.1.16 galactitol 2-dehydrogenase 1.1.1.17 mannitol-1-phosphate 5-dehydrogenase 1.1.1.18 inositol 2-dehydrogenase 1.1.1.19 glucuronate reductase 1.1.1.20 glucuronolactone reductase 1.1.1.21 aldehyde reductase 1.1.1.22 UDP-glucose 6-dehydrogenase 1.1.1.23 histidinol dehydrogenase 1.1.1.24 quinate dehydrogenase 1.1.1.25 shikimate dehydrogenase 1.1.1.26 glyoxylate reductase 1.1.1.27 L-lactate dehydrogenase 1.1.1.28 D-lactate dehydrogenase 1.1.1.29 glycerate dehydrogenase 1.1.1.30 3-hydroxybutyrate dehydrogenase 1.1.1.31 3-hydroxyisobutyrate dehydrogenase 1.1.1.32 mevaldate reductase 1.1.1.33 mevaldate reductase (NADPH) 1.1.1.34 hydroxymethylglutaryl-CoA reductase (NADPH) 1.1.1.35 3-hydroxyacyl-CoA