Patients with PWS and Related Syndromes Display Differentially
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-
Primepcr™Assay Validation Report
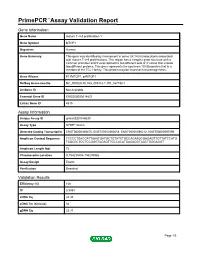
PrimePCR™Assay Validation Report Gene Information Gene Name mature T-cell proliferation 1 Gene Symbol MTCP1 Organism Human Gene Summary This gene was identified by involvement in some t(X;14) translocations associated with mature T-cell proliferations. This region has a complex gene structure with a common promoter and 5' exon spliced to two different sets of 3' exons that encode two different proteins. This gene represents the upstream 13 kDa protein that is a member of the TCL1 family. This protein may be involved in leukemogenesis. Gene Aliases P13MTCP1, p8MTCP1 RefSeq Accession No. NC_000023.10, NG_005114.1, NT_167198.1 UniGene ID Not Available Ensembl Gene ID ENSG00000214827 Entrez Gene ID 4515 Assay Information Unique Assay ID qHsaCED0048630 Assay Type SYBR® Green Detected Coding Transcript(s) ENST00000369476, ENST00000362018, ENST00000596212, ENST00000597096 Amplicon Context Sequence TCCCCTGACCATTAAATGATGCTGTATCTGCCACAAGCGAGAGTTGTTATCCATG TAGCGCTCCTCCGGGTAGAGTTGCCACATGAGAGGTAGCTGGGAGGT Amplicon Length (bp) 72 Chromosome Location X:154293804-154293986 Assay Design Exonic Purification Desalted Validation Results Efficiency (%) 106 R2 0.9994 cDNA Cq 24.34 cDNA Tm (Celsius) 82 gDNA Cq 25.37 Page 1/5 PrimePCR™Assay Validation Report Specificity (%) 100 Information to assist with data interpretation is provided at the end of this report. Page 2/5 PrimePCR™Assay Validation Report MTCP1, Human Amplification Plot Amplification of cDNA generated from 25 ng of universal reference RNA Melt Peak Melt curve analysis of above amplification Standard -

Structural Studies of the Complex Between Akt-In and the Akt2-PH Domain Suggest That the Peptide Acts As an Allosteric Inhibitor of the Akt Kinase
The Open Spectroscopy Journal, 2009, 3, 65-76 65 Open Access Structural Studies of the Complex Between Akt-in and the Akt2-PH Domain Suggest that the Peptide Acts as an Allosteric Inhibitor of the Akt Kinase Virginie Ropars1,2,3, Philippe Barthe1,2,3, Chi-Shien Wang4, Wenlung Chen4, Der-Lii M. Tzou4,5, Anne Descours6, Loïc Martin6, Masayuki Noguchi7, Daniel Auguin1,2,3,,¶ and Christian Roumestand*,1,2,3 1CNRS UMR 5048, Centre de Biochimie Structurale, Montpellier, France 2INSERM U554, Montpellier, France 3Université Montpellier I et II, Montpellier, France 4Department of Applied Chemistry, National Chiayi University, Chiayi 60004, Taiwan, ROC 5Institute of Chemistry, Academia Sinica, Nankang, Taipei 11529, Taiwan, ROC 6CEA, iBiTecs, Service d’Ingénierie Moléculaire des Protéines, 91191 Gif sur Yvette, France 7Division of Cancer Biology, Institute for Genetic Medicine, Hokkaido University, N15 W7, Kita-ku, Sapporo 060-0815, Japan Abstract: Serine/threonine kinase Akt plays a central role in the regulation of cell survival and proliferation. Hence, the search for Akt specific inhibitors constitutes an attractive strategy for anticancer therapy. We have previously demonstrated that the proto-oncogene TCL1 coactivates Akt upon binding to its Plekstrin Homology Domain, and we proposed a model for the structure of the complex TCL1:Akt2-PHD. This model led to the rational design of Akt-in, a peptide inhibitor spanning the A ß-strand of human TCL1 that binds Akt2 PH domain and inhibits the kinase activation. In the present report, we used NMR spectroscopy to determine the 3D structure of the peptide free in solution and bound to Akt2-PHD. -

Protein Interaction Network of Alternatively Spliced Isoforms from Brain Links Genetic Risk Factors for Autism
ARTICLE Received 24 Aug 2013 | Accepted 14 Mar 2014 | Published 11 Apr 2014 DOI: 10.1038/ncomms4650 OPEN Protein interaction network of alternatively spliced isoforms from brain links genetic risk factors for autism Roser Corominas1,*, Xinping Yang2,3,*, Guan Ning Lin1,*, Shuli Kang1,*, Yun Shen2,3, Lila Ghamsari2,3,w, Martin Broly2,3, Maria Rodriguez2,3, Stanley Tam2,3, Shelly A. Trigg2,3,w, Changyu Fan2,3, Song Yi2,3, Murat Tasan4, Irma Lemmens5, Xingyan Kuang6, Nan Zhao6, Dheeraj Malhotra7, Jacob J. Michaelson7,w, Vladimir Vacic8, Michael A. Calderwood2,3, Frederick P. Roth2,3,4, Jan Tavernier5, Steve Horvath9, Kourosh Salehi-Ashtiani2,3,w, Dmitry Korkin6, Jonathan Sebat7, David E. Hill2,3, Tong Hao2,3, Marc Vidal2,3 & Lilia M. Iakoucheva1 Increased risk for autism spectrum disorders (ASD) is attributed to hundreds of genetic loci. The convergence of ASD variants have been investigated using various approaches, including protein interactions extracted from the published literature. However, these datasets are frequently incomplete, carry biases and are limited to interactions of a single splicing isoform, which may not be expressed in the disease-relevant tissue. Here we introduce a new interactome mapping approach by experimentally identifying interactions between brain-expressed alternatively spliced variants of ASD risk factors. The Autism Spliceform Interaction Network reveals that almost half of the detected interactions and about 30% of the newly identified interacting partners represent contribution from splicing variants, emphasizing the importance of isoform networks. Isoform interactions greatly contribute to establishing direct physical connections between proteins from the de novo autism CNVs. Our findings demonstrate the critical role of spliceform networks for translating genetic knowledge into a better understanding of human diseases. -

Replace This with the Actual Title Using All Caps
UNDERSTANDING THE GENETICS UNDERLYING MASTITIS USING A MULTI-PRONGED APPROACH A Dissertation Presented to the Faculty of the Graduate School of Cornell University In Partial Fulfillment of the Requirements for the Degree of Doctor of Philosophy by Asha Marie Miles December 2019 © 2019 Asha Marie Miles UNDERSTANDING THE GENETICS UNDERLYING MASTITIS USING A MULTI-PRONGED APPROACH Asha Marie Miles, Ph. D. Cornell University 2019 This dissertation addresses deficiencies in the existing genetic characterization of mastitis due to granddaughter study designs and selection strategies based primarily on lactation average somatic cell score (SCS). Composite milk samples were collected across 6 sampling periods representing key lactation stages: 0-1 day in milk (DIM), 3- 5 DIM, 10-14 DIM, 50-60 DIM, 90-110 DIM, and 210-230 DIM. Cows were scored for front and rear teat length, width, end shape, and placement, fore udder attachment, udder cleft, udder depth, rear udder height, and rear udder width. Independent multivariable logistic regression models were used to generate odds ratios for elevated SCC (≥ 200,000 cells/ml) and farm-diagnosed clinical mastitis. Within our study cohort, loose fore udder attachment, flat teat ends, low rear udder height, and wide rear teats were associated with increased odds of mastitis. Principal component analysis was performed on these traits to create a single new phenotype describing mastitis susceptibility based on these high-risk phenotypes. Cows (N = 471) were genotyped on the Illumina BovineHD 777K SNP chip and considering all 14 traits of interest, a total of 56 genome-wide associations (GWA) were performed and 28 significantly associated quantitative trait loci (QTL) were identified. -

Kids First Pediatric Research Program (Kids First) Poster Session at ASHG Accelerating Pediatric Genomics Research Through Collaboration October 15Th, 2019
The Gabriella Miller Kids First Pediatric Research Program (Kids First) Poster Session at ASHG Accelerating Pediatric Genomics Research through Collaboration October 15th, 2019 Background The Gabriella Miller Kids First Pediatric Research Program (Kids First) is a trans- NIH Common Fund program initiated in response to the 2014 Gabriella Miller Kids First Research Act. The program’s vision is to alleviate suffering from childhood cancer and structural birth defects by fostering collaborative research to uncover the etiology of these diseases and support data sharing within the pediatric research community. This is implemented through developing the Gabriella Miller Kids First Data Resource (Kids First Data Resource) and populating this resource with whole genome sequence datasets and associated clinical and phenotypic information. Both childhood cancers and structural birth defects are critical and costly conditions associated with substantial morbidity and mortality. Elucidating the underlying genetic etiology of these diseases has the potential to profoundly improve preventative measures, diagnostics, and therapeutic interventions. Purpose During this evening poster session, attendees will gain a broad understanding of the utility of the genomic data generated by Kids First, learn about the progress of Kids First X01 cohort projects, and observe demonstrations of the tools and functionalities of the recently launched Kids First Data Resource Portal. The session is an opportunity for the scientific community and public to engage with Kids First investigators, collaborators, and a growing community of researchers, patient foundations, and families. Several other NIH and external data efforts will present posters and be available to discuss collaboration opportunities as we work together to accelerate pediatric research. -

Parallel Molecular Evolution in Pathways, Genes, and Sites in High-Elevation Hummingbirds Revealed by Comparative Transcriptomics
GBE Parallel Molecular Evolution in Pathways, Genes, and Sites in High-Elevation Hummingbirds Revealed by Comparative Transcriptomics Marisa C.W. Lim1,*, Christopher C. Witt2, Catherine H. Graham1,3,andLilianaM.Davalos 1,4 1Department of Ecology and Evolution, Stony Brook University 2 Museum of Southwestern Biology and Department of Biology, University of New Mexico Downloaded from https://academic.oup.com/gbe/article-abstract/11/6/1552/5494706 by guest on 08 June 2019 3Swiss Federal Research Institute (WSL), Birmensdorf, Switzerland 4Consortium for Inter-Disciplinary Environmental Research, Stony Brook University *Corresponding author: E-mail: [email protected]. Accepted: May 12, 2019 Data deposition: The raw read data have been deposited in the NCBI Sequence Read Archive under BioProject: PRJNA543673, BioSample: SAMN11774663-SAMN11774674, SRA Study: SRP198856. All scripts used for analyses are available on Dryad: doi:10.5061/dryad.v961mb4. Abstract High-elevation organisms experience shared environmental challenges that include low oxygen availability, cold temperatures, and intense ultraviolet radiation. Consequently, repeated evolution of the same genetic mechanisms may occur across high-elevation taxa. To test this prediction, we investigated the extent to which the same biochemical pathways, genes, or sites were subject to parallel molecular evolution for 12 Andean hummingbird species (family: Trochilidae) representing several independent transitions to high elevation across the phylogeny. Across high-elevation species, we discovered parallel evolution for several pathways and genes with evidence of positive selection. In particular, positively selected genes were frequently part of cellular respiration, metabolism, or cell death pathways. To further examine the role of elevation in our analyses, we compared results for low- and high-elevation species and tested different thresholds for defining elevation categories. -

A Computational Approach for Defining a Signature of Β-Cell Golgi Stress in Diabetes Mellitus
Page 1 of 781 Diabetes A Computational Approach for Defining a Signature of β-Cell Golgi Stress in Diabetes Mellitus Robert N. Bone1,6,7, Olufunmilola Oyebamiji2, Sayali Talware2, Sharmila Selvaraj2, Preethi Krishnan3,6, Farooq Syed1,6,7, Huanmei Wu2, Carmella Evans-Molina 1,3,4,5,6,7,8* Departments of 1Pediatrics, 3Medicine, 4Anatomy, Cell Biology & Physiology, 5Biochemistry & Molecular Biology, the 6Center for Diabetes & Metabolic Diseases, and the 7Herman B. Wells Center for Pediatric Research, Indiana University School of Medicine, Indianapolis, IN 46202; 2Department of BioHealth Informatics, Indiana University-Purdue University Indianapolis, Indianapolis, IN, 46202; 8Roudebush VA Medical Center, Indianapolis, IN 46202. *Corresponding Author(s): Carmella Evans-Molina, MD, PhD ([email protected]) Indiana University School of Medicine, 635 Barnhill Drive, MS 2031A, Indianapolis, IN 46202, Telephone: (317) 274-4145, Fax (317) 274-4107 Running Title: Golgi Stress Response in Diabetes Word Count: 4358 Number of Figures: 6 Keywords: Golgi apparatus stress, Islets, β cell, Type 1 diabetes, Type 2 diabetes 1 Diabetes Publish Ahead of Print, published online August 20, 2020 Diabetes Page 2 of 781 ABSTRACT The Golgi apparatus (GA) is an important site of insulin processing and granule maturation, but whether GA organelle dysfunction and GA stress are present in the diabetic β-cell has not been tested. We utilized an informatics-based approach to develop a transcriptional signature of β-cell GA stress using existing RNA sequencing and microarray datasets generated using human islets from donors with diabetes and islets where type 1(T1D) and type 2 diabetes (T2D) had been modeled ex vivo. To narrow our results to GA-specific genes, we applied a filter set of 1,030 genes accepted as GA associated. -

PDF Datasheet
Product Datasheet Cactin Overexpression Lysate NBP2-06542 Unit Size: 0.1 mg Store at -80C. Avoid freeze-thaw cycles. Protocols, Publications, Related Products, Reviews, Research Tools and Images at: www.novusbio.com/NBP2-06542 Updated 3/17/2020 v.20.1 Earn rewards for product reviews and publications. Submit a publication at www.novusbio.com/publications Submit a review at www.novusbio.com/reviews/destination/NBP2-06542 Page 1 of 2 v.20.1 Updated 3/17/2020 NBP2-06542 Cactin Overexpression Lysate Product Information Unit Size 0.1 mg Concentration The exact concentration of the protein of interest cannot be determined for overexpression lysates. Please contact technical support for more information. Storage Store at -80C. Avoid freeze-thaw cycles. Buffer RIPA buffer Target Molecular Weight 88.5 kDa Product Description Description Transient overexpression lysate of chromosome 19 open reading frame 29 (C19orf29), transcript variant 2 The lysate was created in HEK293T cells, using Plasmid ID RC213573 and based on accession number NM_021231. The protein contains a C-MYC/DDK Tag. Gene ID 58509 Gene Symbol CACTIN Species Human Notes HEK293T cells in 10-cm dishes were transiently transfected with a non-lipid polymer transfection reagent specially designed and manufactured for large volume DNA transfection. Transfected cells were cultured for 48hrs before collection. The cells were lysed in modified RIPA buffer (25mM Tris-HCl pH7.6, 150mM NaCl, 1% NP-40, 1mM EDTA, 1xProteinase inhibitor cocktail mix, 1mM PMSF and 1mM Na3VO4, and then centrifuged to clarify the lysate. Protein concentration was measured by BCA protein assay kit.This product is manufactured by and sold under license from OriGene Technologies and its use is limited solely for research purposes. -

Chromatin-Associated Degradation Is Defined by UBXN-3/FAF1 To
ARTICLE Received 27 Jul 2015 | Accepted 5 Jan 2016 | Published 4 Feb 2016 DOI: 10.1038/ncomms10612 OPEN Chromatin-associated degradation is defined by UBXN-3/FAF1 to safeguard DNA replication fork progression Andre´ Franz1, Paul A. Pirson1, Domenic Pilger1,2, Swagata Halder2, Divya Achuthankutty2, Hamid Kashkar3, Kristijan Ramadan2 & Thorsten Hoppe1 The coordinated activity of DNA replication factors is a highly dynamic process that involves ubiquitin-dependent regulation. In this context, the ubiquitin-directed ATPase CDC-48/p97 recently emerged as a key regulator of chromatin-associated degradation in several of the DNA metabolic pathways that assure genome integrity. However, the spatiotemporal control of distinct CDC-48/p97 substrates in the chromatin environment remained unclear. Here, we report that progression of the DNA replication fork is coordinated by UBXN-3/FAF1. UBXN-3/FAF1 binds to the licensing factor CDT-1 and additional ubiquitylated proteins, thus promoting CDC-48/p97-dependent turnover and disassembly of DNA replication factor complexes. Consequently, inactivation of UBXN-3/FAF1 stabilizes CDT-1 and CDC-45/GINS on chromatin, causing severe defects in replication fork dynamics accompanied by pronounced replication stress and eventually resulting in genome instability. Our work identifies a critical substrate selection module of CDC-48/p97 required for chromatin- associated protein degradation in both Caenorhabditis elegans and humans, which is relevant to oncogenesis and aging. 1 Institute for Genetics and CECAD Research Center, University of Cologne, Joseph-Stelzmann-Str. 26, 50931 Cologne, Germany. 2 Department of Oncology, University of Oxford, Cancer Research UK/Medical Research Council Oxford, Institute for Radiation Oncology, Old Road Campus Research Building, OX3 7DQ Oxford, UK. -

Transposon Activation Mutagenesis As a Screening Tool for Identifying
Chen et al. BMC Cancer 2013, 13:93 http://www.biomedcentral.com/1471-2407/13/93 RESEARCH ARTICLE Open Access Transposon activation mutagenesis as a screening tool for identifying resistance to cancer therapeutics Li Chen1,2*, Lynda Stuart2, Toshiro K Ohsumi3, Shawn Burgess4, Gaurav K Varshney4, Anahita Dastur1, Mark Borowsky3, Cyril Benes1, Adam Lacy-Hulbert2 and Emmett V Schmidt2 Abstract Background: The development of resistance to chemotherapies represents a significant barrier to successful cancer treatment. Resistance mechanisms are complex, can involve diverse and often unexpected cellular processes, and can vary with both the underlying genetic lesion and the origin or type of tumor. For these reasons developing experimental strategies that could be used to understand, identify and predict mechanisms of resistance in different malignant cells would be a major advance. Methods: Here we describe a gain-of-function forward genetic approach for identifying mechanisms of resistance. This approach uses a modified piggyBac transposon to generate libraries of mutagenized cells, each containing transposon insertions that randomly activate nearby gene expression. Genes of interest are identified using next- gen high-throughput sequencing and barcode multiplexing is used to reduce experimental cost. Results: Using this approach we successfully identify genes involved in paclitaxel resistance in a variety of cancer cell lines, including the multidrug transporter ABCB1, a previously identified major paclitaxel resistance gene. Analysis of co-occurring transposons integration sites in single cell clone allows for the identification of genes that might act cooperatively to produce drug resistance a level of information not accessible using RNAi or ORF expression screening approaches. Conclusion: We have developed a powerful pipeline to systematically discover drug resistance in mammalian cells in vitro. -

Supplementary Table S4. FGA Co-Expressed Gene List in LUAD
Supplementary Table S4. FGA co-expressed gene list in LUAD tumors Symbol R Locus Description FGG 0.919 4q28 fibrinogen gamma chain FGL1 0.635 8p22 fibrinogen-like 1 SLC7A2 0.536 8p22 solute carrier family 7 (cationic amino acid transporter, y+ system), member 2 DUSP4 0.521 8p12-p11 dual specificity phosphatase 4 HAL 0.51 12q22-q24.1histidine ammonia-lyase PDE4D 0.499 5q12 phosphodiesterase 4D, cAMP-specific FURIN 0.497 15q26.1 furin (paired basic amino acid cleaving enzyme) CPS1 0.49 2q35 carbamoyl-phosphate synthase 1, mitochondrial TESC 0.478 12q24.22 tescalcin INHA 0.465 2q35 inhibin, alpha S100P 0.461 4p16 S100 calcium binding protein P VPS37A 0.447 8p22 vacuolar protein sorting 37 homolog A (S. cerevisiae) SLC16A14 0.447 2q36.3 solute carrier family 16, member 14 PPARGC1A 0.443 4p15.1 peroxisome proliferator-activated receptor gamma, coactivator 1 alpha SIK1 0.435 21q22.3 salt-inducible kinase 1 IRS2 0.434 13q34 insulin receptor substrate 2 RND1 0.433 12q12 Rho family GTPase 1 HGD 0.433 3q13.33 homogentisate 1,2-dioxygenase PTP4A1 0.432 6q12 protein tyrosine phosphatase type IVA, member 1 C8orf4 0.428 8p11.2 chromosome 8 open reading frame 4 DDC 0.427 7p12.2 dopa decarboxylase (aromatic L-amino acid decarboxylase) TACC2 0.427 10q26 transforming, acidic coiled-coil containing protein 2 MUC13 0.422 3q21.2 mucin 13, cell surface associated C5 0.412 9q33-q34 complement component 5 NR4A2 0.412 2q22-q23 nuclear receptor subfamily 4, group A, member 2 EYS 0.411 6q12 eyes shut homolog (Drosophila) GPX2 0.406 14q24.1 glutathione peroxidase -

Analysis of the Indacaterol-Regulated Transcriptome in Human Airway
Supplemental material to this article can be found at: http://jpet.aspetjournals.org/content/suppl/2018/04/13/jpet.118.249292.DC1 1521-0103/366/1/220–236$35.00 https://doi.org/10.1124/jpet.118.249292 THE JOURNAL OF PHARMACOLOGY AND EXPERIMENTAL THERAPEUTICS J Pharmacol Exp Ther 366:220–236, July 2018 Copyright ª 2018 by The American Society for Pharmacology and Experimental Therapeutics Analysis of the Indacaterol-Regulated Transcriptome in Human Airway Epithelial Cells Implicates Gene Expression Changes in the s Adverse and Therapeutic Effects of b2-Adrenoceptor Agonists Dong Yan, Omar Hamed, Taruna Joshi,1 Mahmoud M. Mostafa, Kyla C. Jamieson, Radhika Joshi, Robert Newton, and Mark A. Giembycz Departments of Physiology and Pharmacology (D.Y., O.H., T.J., K.C.J., R.J., M.A.G.) and Cell Biology and Anatomy (M.M.M., R.N.), Snyder Institute for Chronic Diseases, Cumming School of Medicine, University of Calgary, Calgary, Alberta, Canada Received March 22, 2018; accepted April 11, 2018 Downloaded from ABSTRACT The contribution of gene expression changes to the adverse and activity, and positive regulation of neutrophil chemotaxis. The therapeutic effects of b2-adrenoceptor agonists in asthma was general enriched GO term extracellular space was also associ- investigated using human airway epithelial cells as a therapeu- ated with indacaterol-induced genes, and many of those, in- tically relevant target. Operational model-fitting established that cluding CRISPLD2, DMBT1, GAS1, and SOCS3, have putative jpet.aspetjournals.org the long-acting b2-adrenoceptor agonists (LABA) indacaterol, anti-inflammatory, antibacterial, and/or antiviral activity. Numer- salmeterol, formoterol, and picumeterol were full agonists on ous indacaterol-regulated genes were also induced or repressed BEAS-2B cells transfected with a cAMP-response element in BEAS-2B cells and human primary bronchial epithelial cells by reporter but differed in efficacy (indacaterol $ formoterol .