2Nd Quantization(Pdf)
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Part 5: the Bose-Einstein Distribution
PHYS393 – Statistical Physics Part 5: The Bose-Einstein Distribution Distinguishable and indistinguishable particles In the previous parts of this course, we derived the Boltzmann distribution, which described how the number of distinguishable particles in different energy states varied with the energy of those states, at different temperatures: N εj nj = e− kT . (1) Z However, in systems consisting of collections of identical fermions or identical bosons, the wave function of the system has to be either antisymmetric (for fermions) or symmetric (for bosons) under interchange of any two particles. With the allowed wave functions, it is no longer possible to identify a particular particle with a particular energy state. Instead, all the particles are “shared” between the occupied states. The particles are said to be indistinguishable . Statistical Physics 1 Part 5: The Bose-Einstein Distribution Indistinguishable fermions In the case of indistinguishable fermions, the wave function for the overall system must be antisymmetric under the interchange of any two particles. One consequence of this is the Pauli exclusion principle: any given state can be occupied by at most one particle (though we can’t say which particle!) For example, for a system of two fermions, a possible wave function might be: 1 ψ(x1, x 2) = [ψ (x1)ψ (x2) ψ (x2)ψ (x1)] . (2) √2 A B − A B Here, x1 and x2 are the coordinates of the two particles, and A and B are the two occupied states. If we try to put the two particles into the same state, then the wave function vanishes. Statistical Physics 2 Part 5: The Bose-Einstein Distribution Indistinguishable fermions Finding the distribution with the maximum number of microstates for a system of identical fermions leads to the Fermi-Dirac distribution: g n = i . -

The Debye Model of Lattice Heat Capacity
The Debye Model of Lattice Heat Capacity The Debye model of lattice heat capacity is more involved than the relatively simple Einstein model, but it does keep the same basic idea: the internal energy depends on the energy per phonon (ε=ℏΩ) times the ℏΩ/kT average number of phonons (Planck distribution: navg=1/[e -1]) times the number of modes. It is in the number of modes that the difference between the two models occurs. In the Einstein model, we simply assumed that each mode was the same (same frequency, Ω), and that the number of modes was equal to the number of atoms in the lattice. In the Debye model, we assume that each mode has its own K (that is, has its own λ). Since Ω is related to K (by the dispersion relation), each mode has a different Ω. 1. Number of modes In the Debye model we assume that the normal modes consist of STANDING WAVES. If we have travelling waves, they will carry energy through the material; if they contain the heat energy (that will not move) they need to be standing waves. Standing waves are waves that do travel but go back and forth and interfere with each other to create standing waves. Recall that Newton's Second Law gave us atoms that oscillate: [here x = sa where s is an integer and a the lattice spacing] iΩt iKsa us = uo e e ; and if we use the form sin(Ksa) for eiKsa [both indicate oscillations]: iΩt us = uo e sin(Ksa) . We now apply the boundary conditions on our solution: to have standing waves both ends of the wave must be tied down, that is, we must have u0 = 0 and uN = 0. -

Competition Between Paramagnetism and Diamagnetism in Charged
Competition between paramagnetism and diamagnetism in charged Fermi gases Xiaoling Jian, Jihong Qin, and Qiang Gu∗ Department of Physics, University of Science and Technology Beijing, Beijing 100083, China (Dated: November 10, 2018) The charged Fermi gas with a small Lande-factor g is expected to be diamagnetic, while that with a larger g could be paramagnetic. We calculate the critical value of the g-factor which separates the dia- and para-magnetic regions. In the weak-field limit, gc has the same value both at high and low temperatures, gc = 1/√12. Nevertheless, gc increases with the temperature reducing in finite magnetic fields. We also compare the gc value of Fermi gases with those of Boltzmann and Bose gases, supposing the particle has three Zeeman levels σ = 1, 0, and find that gc of Bose and Fermi gases is larger and smaller than that of Boltzmann gases,± respectively. PACS numbers: 05.30.Fk, 51.60.+a, 75.10.Lp, 75.20.-g I. INTRODUCTION mK and 10 µK, respectively. The ions can be regarded as charged Bose or Fermi gases. Once the quantum gas Magnetism of electron gases has been considerably has both the spin and charge degrees of freedom, there studied in condensed matter physics. In magnetic field, arises the competition between paramagnetism and dia- the magnetization of a free electron gas consists of two magnetism, as in electrons. Different from electrons, the independent parts. The spin magnetic moment of elec- g-factor for different magnetic ions is diverse, ranging trons results in the paramagnetic part (the Pauli para- from 0 to 2. -

Phys 446: Solid State Physics / Optical Properties Lattice Vibrations
Solid State Physics Lecture 5 Last week: Phys 446: (Ch. 3) • Phonons Solid State Physics / Optical Properties • Today: Einstein and Debye models for thermal capacity Lattice vibrations: Thermal conductivity Thermal, acoustic, and optical properties HW2 discussion Fall 2007 Lecture 5 Andrei Sirenko, NJIT 1 2 Material to be included in the test •Factors affecting the diffraction amplitude: Oct. 12th 2007 Atomic scattering factor (form factor): f = n(r)ei∆k⋅rl d 3r reflects distribution of electronic cloud. a ∫ r • Crystalline structures. 0 sin()∆k ⋅r In case of spherical distribution f = 4πr 2n(r) dr 7 crystal systems and 14 Bravais lattices a ∫ n 0 ∆k ⋅r • Crystallographic directions dhkl = 2 2 2 1 2 ⎛ h k l ⎞ 2πi(hu j +kv j +lw j ) and Miller indices ⎜ + + ⎟ •Structure factor F = f e ⎜ a2 b2 c2 ⎟ ∑ aj ⎝ ⎠ j • Definition of reciprocal lattice vectors: •Elastic stiffness and compliance. Strain and stress: definitions and relation between them in a linear regime (Hooke's law): σ ij = ∑Cijklε kl ε ij = ∑ Sijklσ kl • What is Brillouin zone kl kl 2 2 C •Elastic wave equation: ∂ u C ∂ u eff • Bragg formula: 2d·sinθ = mλ ; ∆k = G = eff x sound velocity v = ∂t 2 ρ ∂x2 ρ 3 4 • Lattice vibrations: acoustic and optical branches Summary of the Last Lecture In three-dimensional lattice with s atoms per unit cell there are Elastic properties – crystal is considered as continuous anisotropic 3s phonon branches: 3 acoustic, 3s - 3 optical medium • Phonon - the quantum of lattice vibration. Elastic stiffness and compliance tensors relate the strain and the Energy ħω; momentum ħq stress in a linear region (small displacements, harmonic potential) • Concept of the phonon density of states Hooke's law: σ ij = ∑Cijklε kl ε ij = ∑ Sijklσ kl • Einstein and Debye models for lattice heat capacity. -

Solid State Physics 2 Lecture 5: Electron Liquid
Physics 7450: Solid State Physics 2 Lecture 5: Electron liquid Leo Radzihovsky (Dated: 10 March, 2015) Abstract In these lectures, we will study itinerate electron liquid, namely metals. We will begin by re- viewing properties of noninteracting electron gas, developing its Greens functions, analyzing its thermodynamics, Pauli paramagnetism and Landau diamagnetism. We will recall how its thermo- dynamics is qualitatively distinct from that of a Boltzmann and Bose gases. As emphasized by Sommerfeld (1928), these qualitative di↵erence are due to the Pauli principle of electons’ fermionic statistics. We will then include e↵ects of Coulomb interaction, treating it in Hartree and Hartree- Fock approximation, computing the ground state energy and screening. We will then study itinerate Stoner ferromagnetism as well as various response functions, such as compressibility and conduc- tivity, and screening (Thomas-Fermi, Debye). We will then discuss Landau Fermi-liquid theory, which will allow us understand why despite strong electron-electron interactions, nevertheless much of the phenomenology of a Fermi gas extends to a Fermi liquid. We will conclude with discussion of electrons on the lattice, treated within the Hubbard and t-J models and will study transition to a Mott insulator and magnetism 1 I. INTRODUCTION A. Outline electron gas ground state and excitations • thermodynamics • Pauli paramagnetism • Landau diamagnetism • Hartree-Fock theory of interactions: ground state energy • Stoner ferromagnetic instability • response functions • Landau Fermi-liquid theory • electrons on the lattice: Hubbard and t-J models • Mott insulators and magnetism • B. Background In these lectures, we will study itinerate electron liquid, namely metals. In principle a fully quantum mechanical, strongly Coulomb-interacting description is required. -

Lecture Notes
Solid State Physics PHYS 40352 by Mike Godfrey Spring 2012 Last changed on May 22, 2017 ii Contents Preface v 1 Crystal structure 1 1.1 Lattice and basis . .1 1.1.1 Unit cells . .2 1.1.2 Crystal symmetry . .3 1.1.3 Two-dimensional lattices . .4 1.1.4 Three-dimensional lattices . .7 1.1.5 Some cubic crystal structures ................................ 10 1.2 X-ray crystallography . 11 1.2.1 Diffraction by a crystal . 11 1.2.2 The reciprocal lattice . 12 1.2.3 Reciprocal lattice vectors and lattice planes . 13 1.2.4 The Bragg construction . 14 1.2.5 Structure factor . 15 1.2.6 Further geometry of diffraction . 17 2 Electrons in crystals 19 2.1 Summary of free-electron theory, etc. 19 2.2 Electrons in a periodic potential . 19 2.2.1 Bloch’s theorem . 19 2.2.2 Brillouin zones . 21 2.2.3 Schrodinger’s¨ equation in k-space . 22 2.2.4 Weak periodic potential: Nearly-free electrons . 23 2.2.5 Metals and insulators . 25 2.2.6 Band overlap in a nearly-free-electron divalent metal . 26 2.2.7 Tight-binding method . 29 2.3 Semiclassical dynamics of Bloch electrons . 32 2.3.1 Electron velocities . 33 2.3.2 Motion in an applied field . 33 2.3.3 Effective mass of an electron . 34 2.4 Free-electron bands and crystal structure . 35 2.4.1 Construction of the reciprocal lattice for FCC . 35 2.4.2 Group IV elements: Jones theory . 36 2.4.3 Binding energy of metals . -

Intermediate Statistics in Thermoelectric Properties of Solids
Intermediate statistics in thermoelectric properties of solids André A. Marinho1, Francisco A. Brito1,2 1 Departamento de Física, Universidade Federal de Campina Grande, 58109-970 Campina Grande, Paraíba, Brazil and 2 Departamento de Física, Universidade Federal da Paraíba, Caixa Postal 5008, 58051-970 João Pessoa, Paraíba, Brazil (Dated: July 23, 2019) Abstract We study the thermodynamics of a crystalline solid by applying intermediate statistics manifested by q-deformation. We based part of our study on both Einstein and Debye models, exploring primarily de- formed thermal and electrical conductivities as a function of the deformed Debye specific heat. The results revealed that the q-deformation acts in two different ways but not necessarily as independent mechanisms. It acts as a factor of disorder or impurity, modifying the characteristics of a crystalline structure, which are phenomena described by q-bosons, and also as a manifestation of intermediate statistics, the B-anyons (or B-type systems). For the latter case, we have identified the Schottky effect, normally associated with high-Tc superconductors in the presence of rare-earth-ion impurities, and also the increasing of the specific heat of the solids beyond the Dulong-Petit limit at high temperature, usually related to anharmonicity of interatomic interactions. Alternatively, since in the q-bosons the statistics are in principle maintained the effect of the deformation acts more slowly due to a small change in the crystal lattice. On the other hand, B-anyons that belong to modified statistics are more sensitive to the deformation. PACS numbers: 02.20-Uw, 05.30-d, 75.20-g arXiv:1907.09055v1 [cond-mat.stat-mech] 21 Jul 2019 1 I. -
![Arxiv:1608.06287V1 [Hep-Th] 22 Aug 2016 Published in Int](https://docslib.b-cdn.net/cover/7243/arxiv-1608-06287v1-hep-th-22-aug-2016-published-in-int-487243.webp)
Arxiv:1608.06287V1 [Hep-Th] 22 Aug 2016 Published in Int
Preprint typeset in JHEP style - HYPER VERSION Surprises with Nonrelativistic Naturalness Petr Hoˇrava Berkeley Center for Theoretical Physics and Department of Physics University of California, Berkeley, CA 94720-7300, USA and Theoretical Physics Group, Lawrence Berkeley National Laboratory Berkeley, CA 94720-8162, USA Abstract: We explore the landscape of technical naturalness for nonrelativistic systems, finding surprises which challenge and enrich our relativistic intuition already in the simplest case of a single scalar field. While the immediate applications are expected in condensed matter and perhaps in cosmology, the study is motivated by the leading puzzles of funda- mental physics involving gravity: The cosmological constant problem and the Higgs mass hierarchy problem. This brief review is based on talks and lectures given at the 2nd LeCosPA Symposium on Everything About Gravity at National Taiwan University, Taipei, Taiwan (December 2015), to appear in the Proceedings; at the International Conference on Gravitation and Cosmology, KITPC, Chinese Academy of Sciences, Beijing, China (May 2015); at the Symposium Celebrating 100 Years of General Relativity, Guanajuato, Mexico (November 2015); and at the 54. Internationale Universit¨atswochen f¨urTheoretische Physik, Schladming, Austria (February 2016). arXiv:1608.06287v1 [hep-th] 22 Aug 2016 Published in Int. J. Mod. Phys. D Contents 1. Puzzles of Naturalness 1 1.1 Technical Naturalness 2 2. Naturalness in Nonrelativistic Systems 3 2.1 Towards the Classification of Nonrelativistic Nambu-Goldstone Modes 3 2.2 Polynomial Shift Symmetries 5 2.3 Naturalness of Cascading Hierarchies 5 3. Interacting Theories with Polynomial Shift Symmetries 6 3.1 Invariants of Polynomial Shift Symmetries 6 3.2 Polynomial Shift Invariants and Graph Theory 6 3.3 Examples 7 4. -

Energy Bands in Crystals
Energy Bands in Crystals This chapter will apply quantum mechanics to a one dimensional, periodic lattice of potential wells which serves as an analogy to electrons interacting with the atoms of a crystal. We will show that as the number of wells becomes large, the allowed energy levels for the electron form nearly continuous energy bands separated by band gaps where no electron can be found. We thus have an interesting quantum system which exhibits many dual features of the quantum continuum and discrete spectrum. several tenths nm The energy band structure plays a crucial role in the theory of electron con- ductivity in the solid state and explains why materials can be classified as in- sulators, conductors and semiconductors. The energy band structure present in a semiconductor is a crucial ingredient in understanding how semiconductor devices work. Energy levels of “Molecules” By a “molecule” a quantum system consisting of a few periodic potential wells. We have already considered a two well molecular analogy in our discussions of the ammonia clock. 1 V -c sin(k(x-b)) V -c sin(k(x-b)) cosh βx sinhβ x V=0 V = 0 0 a b d 0 a b d k = 2 m E β= 2m(V-E) h h Recall for reasonably large hump potentials V , the two lowest lying states (a even ground state and odd first excited state) are very close in energy. As V →∞, both the ground and first excited state wave functions become completely isolated within a well with little wave function penetration into the classically forbidden central hump. -
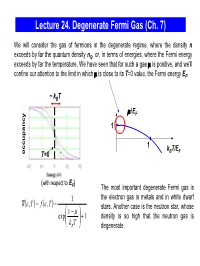
Lecture 24. Degenerate Fermi Gas (Ch
Lecture 24. Degenerate Fermi Gas (Ch. 7) We will consider the gas of fermions in the degenerate regime, where the density n exceeds by far the quantum density nQ, or, in terms of energies, where the Fermi energy exceeds by far the temperature. We have seen that for such a gas μ is positive, and we’ll confine our attention to the limit in which μ is close to its T=0 value, the Fermi energy EF. ~ kBT μ/EF 1 1 kBT/EF occupancy T=0 (with respect to E ) F The most important degenerate Fermi gas is 1 the electron gas in metals and in white dwarf nε()(),, T= f ε T = stars. Another case is the neutron star, whose ε⎛ − μ⎞ exp⎜ ⎟ +1 density is so high that the neutron gas is ⎝kB T⎠ degenerate. Degenerate Fermi Gas in Metals empty states ε We consider the mobile electrons in the conduction EF conduction band which can participate in the charge transport. The band energy is measured from the bottom of the conduction 0 band. When the metal atoms are brought together, valence their outer electrons break away and can move freely band through the solid. In good metals with the concentration ~ 1 electron/ion, the density of electrons in the electron states electron states conduction band n ~ 1 electron per (0.2 nm)3 ~ 1029 in an isolated in metal electrons/m3 . atom The electrons are prevented from escaping from the metal by the net Coulomb attraction to the positive ions; the energy required for an electron to escape (the work function) is typically a few eV. -

Chapter 13 Ideal Fermi
Chapter 13 Ideal Fermi gas The properties of an ideal Fermi gas are strongly determined by the Pauli principle. We shall consider the limit: k T µ,βµ 1, B � � which defines the degenerate Fermi gas. In this limit, the quantum mechanical nature of the system becomes especially important, and the system has little to do with the classical ideal gas. Since this chapter is devoted to fermions, we shall omit in the following the subscript ( ) that we used for the fermionic statistical quantities in the previous chapter. − 13.1 Equation of state Consider a gas ofN non-interacting fermions, e.g., electrons, whose one-particle wave- functionsϕ r(�r) are plane-waves. In this case, a complete set of quantum numbersr is given, for instance, by the three cartesian components of the wave vector �k and thez spin projectionm s of an electron: r (k , k , k , m ). ≡ x y z s Spin-independent Hamiltonians. We will consider only spin independent Hamiltonian operator of the type ˆ 3 H= �k ck† ck + d r V(r)c r†cr , �k � where thefirst and the second terms are respectively the kinetic and th potential energy. The summation over the statesr (whenever it has to be performed) can then be reduced to the summation over states with different wavevectork(p=¯hk): ... (2s + 1) ..., ⇒ r � �k where the summation over the spin quantum numberm s = s, s+1, . , s has been taken into account by the prefactor (2s + 1). − − 159 160 CHAPTER 13. IDEAL FERMI GAS Wavefunctions in a box. We as- sume that the electrons are in a vol- ume defined by a cube with sidesL x, Ly,L z and volumeV=L xLyLz. -

Distortion Correction and Momentum Representation of Angle-Resolved
Distortion Correction and Momentum Representation of Angle-Resolved Photoemission Data Jonathan Adam Rosen 13.Sc., University of California at Santa Cruz, 2006 A THESIS SUBMIYI’ED 1N PARTIAL FULFILLMENT OF THE REQUIREMENTS FOR THE DEGREE OF MASTER OF SCIENCE in THE FACULTY OF GRADUATE STUDIES (Physics) THE UNIVERSITY OF BRETISH COLUMBIA (Vancouver) October 2008 © Jonathan Adam Rosen, 2008 Abstract Angle Resolve Photoemission Spectroscopy (ARPES) experiments provides a map of intensity as function of angles and electron kinetic energy to measure the many-body spectral function, but the raw data returned by standard apparatus is not ready for analysis. An image warping based distortion correction from slit array calibration is shown to provide the relevant information for construction of ARPES intensity as a function of electron momentum. A theory is developed to understand the calculation and uncertainty of the distortion corrected angle space data and the final momentum data. An experimental procedure for determination of the electron analyzer focal point is described and shown to be in good agreement with predictions. The electron analyzer at the Quantum Materials Laboratory at UBC is found to have a focal point at cryostat position 1.09mm within 1.00 mm, and the systematic error in the angle is found to be 0.2 degrees. The angular error is shown to be proportional to a functional form of systematic error in the final ARPES data that is highly momentum dependent. 11 Table .of Contents Abstract ii Table of Contents iii List of Tables v