Hepatitis C Virus and Pegiviruses Identification of Rodent Homologs Of
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

GB Virus C/Hepatitis G Virus Does Not Induce Expression of P44 Antigen in Chimpanzee Hepatocytes Yohko K
Jpn. J. Infect. Dis., 54, 2001 and skin were removed was purchased on the previous night ongln Of infection was not clear. The food service in a festival of the festival. It was left in the room temperature ovemight. as described in the present case does not requlre regulation The cooking started at 4 0'clock in the momlng by roastlng under the food hyglene law. This incident indicated the the bulk meat by rotation over a gas bumer. The meat was necessity of proper measures for food security in this type of covered by a large steel box during roastlng. It was cut into short temporary food service and regulatory measures in case small pleCeS and served at 10 o'clock in the momlng. Some of possible occurrences of food poISOnlng. noted that some portion of the meat was rare; i.e., it was insufficiently cooked. Laboratory and other epidemiologlCal data will be published ln the outbreak, a total 58 cases were counted. There were in Infectious Agents Surveillance Report, vol. 22 (June, 200 I ). 41 primary infections, 1 1 secondary infections including a We thank the clinical institutions, schools and other institu- case which took place in a nursery school, and 6 cases whose tions fわr their collaboration. Laboratory and Epidemiology Communications GB Virus C/Hepatitis G Virus Does Not Induce Expression of p44 Antigen in Chimpanzee Hepatocytes Yohko K. Shimizu*, Minako Hijikata and Hiroshi Yoshikural Department ofRespiratoyy Diseases, Research Institute, International Medical Center ofJapan, Toyama 1-21-1 Shinjuku-ku, Too,0 162-8655 and JNational Institute ofInfectious Diseases, Toyama 1-23-1, Shinjuku-ku, Tokyo 162-8640 Communicated by Hiroshi Ybshikura (Accepted June 1, 2001) The cytoplasmic antlgen, p44, was orlglnally discovered found chimpanzees whose sera were positive for GBV-C什IGV in hepatocytes of chimpanzees experimentally infected with RNA by RTIPCR. -

Discovery of a Novel Simian Pegivirus in Common Marmosets
bioRxiv preprint doi: https://doi.org/10.1101/2020.05.24.113662; this version posted May 24, 2020. The copyright holder for this preprint (which was not certified by peer review) is the author/funder, who has granted bioRxiv a license to display the preprint in perpetuity. It is made available under aCC-BY-NC 4.0 International license. 1 Discovery of a novel simian pegivirus in 2 common marmosets (Callithrix jacchus) 3 with lymphocytic enteritis 4 5 Heffron AS1, Lauck M1, Somsen ED1, Townsend EC1, Bailey AL2, Eickhoff J3, Newman CM1, 6 Kuhn J4, Mejia A5, Simmons HA5, O'Connor DH1. 7 Affiliations 8 1 Department of Pathology and Laboratory Medicine, School of Medicine and Public Health, 9 University of Wisconsin-Madison, Madison, Wisconsin, United States of America 10 2 Department of Pathology and Immunology, Washington University School of Medicine, St. 11 Louis, Missouri, United States of America 12 3 Department of Biostatistics & Medical Informatics, University of Wisconsin-Madison, Madison, 13 WI, United States of America 14 4 Integrated Research Facility at Fort Detrick, National Institute of Allergy and Infectious 15 Diseases, National Institutes of Health, Fort Detrick, Frederick, Maryland, United States of 16 America 17 5 Wisconsin National Primate Research Center, University of Wisconsin-Madison, Madison, 18 Wisconsin, United States of America 19 Abstract 20 From 2010 to 2015, 73 common marmosets (Callithrix jacchus) housed at the Wisconsin 21 National Primate Research Center (WNPRC) were diagnosed postmortem with lymphocytic 22 enteritis. We used unbiased deep-sequencing to screen the blood of deceased enteritis-positive 23 marmosets for the presence of RNA viruses. -

Prevalence and Clinical Significance of HGV/GBV-C Infection in Patients
Jpn. J. Infect. Dis., 59, 25-30, 2006 Original Article Prevalence and Clinical Significance of HGV/GBV-C Infection in Patients with Chronic Hepatitis B or C Jeng-Fu Yang1,2, Chia-Yen Dai2,3, Wan-Long Chuang2, Wen-Yi Lin1,2, Zu-Yau Lin2, Shinn-Cherng Chen2, Ming-Yuh Hsieh2, Liang-Yen Wang2, Jung-Fa Tsai2, Wen-Yu Chang2 and Ming-Lung Yu1,2* 1Department of Preventive Medicine and 2Department of Medicine, Kaohsiung Medical University Hospital, and 3Department of Occupational Medicine, Kaohsiung Municipal HsiaoKang Hospital, Kaohsiung, Taiwan (Received July 5, 2005. Accepted November 11, 2005) SUMMARY: Hepatitis G virus/GB virus-C (HGV/GBV-C) is a newly identified Flavivirus. Its clinical signifi- cance in chronic hepatitis B and C remains controversial. Infection with HGV/GBV-C was surveyed in 500 blood donors, 130 patients with chronic hepatitis B and 173 with hepatitis C, with chronic liver disease, cirrhosis, and/or hepatocellular carcinoma (HCC). HGV/GBV-C RNA was detected by reverse transcription-polymerase chain reaction. An antibody to HGV/GBV-C’s second envelope protein (anti-E2 Ab) was detected using an enzyme immunoassay. The prevalence of HGV/GBV-C RNA was 3.4% and the exposure rate 10.2% in blood donors. The prevalence of HGV/GBV-C RNA in patients with chronic hepatitis B and hepatitis C was 7.7 and 17.3%, respectively (P = 0.002). The prevalence of the HGV/GBV-C infection in hepatitis B carriers increased with the severity of chronic liver disease and risk of HCC. The age and duration of hepatitis B virus infection were the more important contributing factors. -

Metagenomic Identification of Diverse Animal Hepaciviruses and Pegiviruses
bioRxiv preprint doi: https://doi.org/10.1101/2020.05.16.100149; this version posted May 17, 2020. The copyright holder for this preprint (which was not certified by peer review) is the author/funder, who has granted bioRxiv a license to display the preprint in perpetuity. It is made available under aCC-BY-NC-ND 4.0 International license. 1 Metagenomic identification of diverse animal hepaciviruses and 2 pegiviruses 3 4 5 Ashleigh F. Porter1, John H.-O. Pettersson1,2, Wei-Shan Chang1, Erin Harvey1, Karrie Rose3, 6 Mang Shi5, John-Sebastian Eden1,4, Jan Buchmann1, Craig Moritz6, Edward C. Holmes1 7 8 9 1. Marie Bashir Institute for Infectious Diseases and Biosecurity, School of Life and 10 Environmental Sciences and School of Medical Sciences, The University of Sydney, 11 Sydney, Australia. 12 2. Zoonosis Science Center, Department of Medical Biochemistry and Microbiology, 13 Uppsala University, Uppsala, Sweden. 14 3. Australian Registry of Wildlife Health, Taronga Conservation Society Australia, 15 Mosman, Australia. 16 4. Centre for Virus Research, Westmead Institute for Medical Research, Westmead, 17 Australia. 18 5. School of Medicine, Sun Yat-sen University, Guangzhou, China. 19 6. Research School of Biology, and Centre for Biodiversity Analysis, Australian National 20 University, Acton, ACT, Australia. 21 22 23 Address correspondence to: 24 Edward C. Holmes, 25 Marie Bashir Institute for Infectious Diseases and Biosecurity, School of Life and 26 Environmental Sciences and School of Medical Sciences, The University of Sydney, 27 Sydney, Australia 28 [email protected] 1 bioRxiv preprint doi: https://doi.org/10.1101/2020.05.16.100149; this version posted May 17, 2020. -

The GB Viruses: a Review and Proposed Classification of GBV-A, GBV-C (HGV), and GBV-D in Genus Pegivirus Within the Family Flaviviridae
Journal of General Virology (2011), 92, 233–246 DOI 10.1099/vir.0.027490-0 Review The GB viruses: a review and proposed classification of GBV-A, GBV-C (HGV), and GBV-D in genus Pegivirus within the family Flaviviridae Jack T. Stapleton,1 Steven Foung,2 A. Scott Muerhoff,3 Jens Bukh4,5 and Peter Simmonds5 Correspondence 1Department of Internal Medicine, Veterans Administration Medical Center and the University Jack T. Stapleton of Iowa, Iowa City, IA, USA [email protected] 2Department of Pathology, Stanford University, Stanford, CA, USA 3Abbott Diagnostics Division, Abbott Laboratories, Abbott Park, IL, USA 4Copenhagen Hepatitis C Program (CO-HEP), Department of Infectious Diseases and Clinical Research Centre, Copenhagen University Hospital, Hvidovre, Denmark, and Department of International Health, Immunology and Microbiology, Faculty of Health Sciences, University of Copenhagen, Denmark 5Centre for Infectious Diseases, University of Edinburgh, Edinburgh, UK In 1967, it was reported that experimental inoculation of serum from a surgeon (G.B.) with acute hepatitis into tamarins resulted in hepatitis. In 1995, two new members of the family Flaviviridae, named GBV-A and GBV-B, were identified in tamarins that developed hepatitis following inoculation with the 11th GB passage. Neither virus infects humans, and a number of GBV-A variants were identified in wild New World monkeys that were captured. Subsequently, a related human virus was identified [named GBV-C or hepatitis G virus (HGV)], and recently a more distantly related virus (named GBV-D) was discovered in bats. Only GBV-B, a second species within the genus Hepacivirus (type species hepatitis C virus), has been shown to cause hepatitis; it causes acute hepatitis in experimentally infected tamarins. -
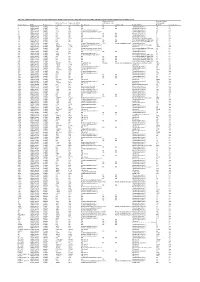
Table S4. Comparison Between the Discvr Results and the BLAST
Table S4. Comparison between the DisCVR results and the BLAST results from the original manuscript for healthy patients and patients with unexplained acute febrile illness Number of k-mers Number of reads Number of k-mers Number of distinct matching the 2nd matching with Sample_Name RUN Patient status matching k-mers matching Top Virus hits hit 2nd Virus Hit Top BLAST hit BLAST Concordance 3 SRR1748140 healthy NA NA NA NA NA Human.adenovirus.C 10 no 4 SRR1748141 healthy NA NA NA NA NA Human.adenovirus.C 65 no 8 SRR1748143 healthy 943 379 Human mastadenovirus C Human.adenovirus.C 65 yes 10 SRR1748147 healthy 1669 133 Human immunodeficiency virus 1 Human.adenovirus.C 26 no 14 SRR1748151 healthy NA NA NA NA NA Human.adenovirus.C 4 no 15 SRR1748152 healthy NA NA NA NA NA Human.adenovirus.C 6 no 108 SRR1748162 healthy NA NA NA NA NA Human.adenovirus.C 6 no 137 SRR1748172 healthy 15343 4714 Human immunodeficiency virus 1 Human.immunodeficiency.virus 314 yes 173 SRR1748173 healthy NA NA NA NA NA Heterosigma.akashiwo.RNA.virus 20 no 245 SRR1748177 healthy NA NA NA NA NA Heterosigma.akashiwo.RNA.virus 17 no 312 SRR1748178 healthy 2843 4 Human T-lymphotropic virus 1 1282 Human mastadenovirus C Human.adenovirus.C 70 no 316 SRR1748179 healthy 22915 5941 Human immunodeficiency virus 1 Human.immunodeficiency.virus 342 yes 319 SRR1748180 healthy 2247683 21201 GB virus C GB.virus.C 20629 yes 325 SRR1748181 healthy 1006 177 Human immunodeficiency virus 1 Heterosigma.akashiwo.RNA.virus 60 no 329 SRR1748182 healthy 1513 124 Human immunodeficiency virus 1 -

GB/Hepatitis G Viruses Likelihood of Secondary Transmission: • Probably Frequent, Based on the Prevalence of Virus in Disease Agent: Blood Donors
APPENDIX 2 GB/Hepatitis G Viruses Likelihood of Secondary Transmission: • Probably frequent, based on the prevalence of virus in Disease Agent: blood donors. • GB viruses (GBV-C/HGV is an acronym for GB virus-C At-Risk Populations: and hepatitis G virus strain variants.) • Blood recipients, injection-drug users, and infants Disease Agent Characteristics: born to infected mothers • Family: Flaviviridae; Genus: Unclassified for GBV-C Vector and Reservoir Involved: • Virion morphology and size: Enveloped, nucleo- capsid of unknown symmetry, 50-100 nm in diameter • Humans • Nucleic acid: Linear, positive-sense, single-stranded Blood Phase: RNA, ~9.4 kb in length • Physicochemical properties: Less stable in CsCl than • Viremic phase can last from weeks to years HCV; other properties not established for these Survival/Persistence in Blood Products: viruses, but, under in vitro conditions, other flavivi- ruses are stable in alkaline environment of pH 8 and • Survives refrigeration are sensitive to treatment with heat, organic solvents, • Inactivated by solvent-detergent and detergents. Transmission by Blood Transfusion: Disease Name: • Well documented in prospective studies • No known disease association Cases/Frequency in Population: Priority Level: • The prevalence of viremia is 1-4%, and antibody • Scientific/Epidemiologic evidence regarding blood prevalence is 3-14% in blood donors. safety: Absent; transmission documented, but no • Prevalence of 10-20% in patients with viral and non- disease associated despite extensive studies viral liver diseases based on antibody and RNA • Public perception and/or regulatory concern regard- • Prevalence of 75-90% in injection-drug users (anti- ing blood safety: Absent body and RNA) • Public concern regarding disease agent: Absent Incubation Period: Background: • Viremia becomes detectable from 2 days to 2 weeks • In the 1960s, serum from a surgeon (GB) with acute postexposure. -

Human Pegivirus Isolates Characterized by Deep Sequencing
Human pegivirus isolates characterized by deep sequencing from hepatitis C virus-RNA and human immunodeficiency virus-RNA–positive blood donations, France François Jordier, Marie-Laurence Deligny, Romain Barré, Catherine Robert, Vital Galicher, Rathviro Uch, Pierre-Edouard Fournier, Didier Raoult, Philippe Biagini To cite this version: François Jordier, Marie-Laurence Deligny, Romain Barré, Catherine Robert, Vital Galicher, et al.. Human pegivirus isolates characterized by deep sequencing from hepatitis C virus-RNA and human immunodeficiency virus-RNA–positive blood donations, France. Journal of Medical Virology, Wiley- Blackwell, 2018, 91 (1), pp.38-44. 10.1002/jmv.25290. hal-01869364 HAL Id: hal-01869364 https://hal.archives-ouvertes.fr/hal-01869364 Submitted on 27 Jul 2021 HAL is a multi-disciplinary open access L’archive ouverte pluridisciplinaire HAL, est archive for the deposit and dissemination of sci- destinée au dépôt et à la diffusion de documents entific research documents, whether they are pub- scientifiques de niveau recherche, publiés ou non, lished or not. The documents may come from émanant des établissements d’enseignement et de teaching and research institutions in France or recherche français ou étrangers, des laboratoires abroad, or from public or private research centers. publics ou privés. Copyright Human pegivirus isolates characterized by deep sequencing from hepatitis C virus‐RNA and human immunodeficiency virus‐RNA–positive blood donations, France François Jordier1 | Marie‐Laurence Deligny1 | Romain Barré1 | Catherine Robert2 | Vital Galicher1 | Rathviro Uch1 | Pierre‐Edouard Fournier3 | Didier Raoult2 | Philippe Biagini1 1Biologie des Groupes Sanguins, ‐ Etablissement Français du Sang Provence Human pegivirus (HPgV, formerly GBV C) is a member of the genus Pegivirus, family Alpes Côte d’Azur Corse, Aix Marseille Flaviviridae. -

8.3 Notebook Annotated
Hans L. Tillmann, md Department of Gastroenterology, When Two Infections Hepatology, and Endocrinology Diagnostic Laboratory for Viral and Autoimmune Hepatitis Are Better Than One: Medizinische Hochshule Hannover, Germany The Exceptional Role of Summary by Tim Horn Edited by Douglas Dieterich, md, and GB Virus-C (GBV-C) in Stephen Locarnini, mbbs, phd, frc (path) HIV Disease Reprinted from The prn Notebook® | september 2003 | Dr. James F. Braun, Editor-in-Chief | Tim Horn, Executive Editor. | Published in New York City by the Physicians’ Research Network, Inc.® John Graham Brown, Executive Director | For further information and other articles available online, visit http://www.prn.org | All rights reserved. ©september 2003 when it comes to hiv and hepatitis virus coinfections, the pages fected with hcv (Linnen, 1996). A second virus was isolated in this pa- of The prn Notebook have been filled with numerous reports high- tient, which the Genelabs team called the hepatitis G virus (hgv). lighting the distressing prevalence and negative consequences of both With the discovery of both viruses, researchers set out to deter- hepatitis B virus (hbv) and hepatitis C virus (hcv) in hiv-infected in- mine the full genomic sequences of the isolates. It turned out that dividuals. However, it does not appear that all coinfections are harmful. gbv-c and hgv are closely related isolates of the same virus, with more In fact, some may be associated with a significant survival advantage— than 95% sequence homology (Alter, 1996). It was also determined a theory that has many hiv experts both perplexed and excited. that the genomic organization of these viruses is similar to that of hcv gb virus-c (gbv-c) infection is one such example. -

Dissertation Bats As Reservoir Hosts
DISSERTATION BATS AS RESERVOIR HOSTS: EXPLORING NOVEL VIRUSES IN NEW WORLD BATS Submitted by Ashley Malmlov Department of Microbiology, Immunology, and Pathology In partial fulfillment of the requirements For the Degree of Doctor of Philosophy Colorado State University Fort Collins, Colorado Spring 2018 Doctoral Committee: Advisor: Tony Schountz Richard Bowen Page Dinsmore Kristy Pabilonia Copyright by Ashley Malmlov 2018 All Rights Reserved ABSTRACT BATS AS RESERVOIR HOSTS: EXPLORING NOVEL VIRUSES IN NEW WORLD BATS Order Chiroptera is oft incriminated for their capacity to serve as reservoirs for many high profile human pathogens, including Ebola virus, Marburg virus, severe acute respiratory syndrome coronavirus, Nipah virus and Hendra virus. Additionally, bats are postulated to be the original hosts for such virus families and subfamilies as Paramyxoviridae and Coronavirinae. Given the perceived risk bats may impart upon public health, numerous explorations have been done to delineate if in fact bats do host more viruses than other animal species, such as rodents, and to ascertain what is unique about bats to allow them to maintain commensal relationships with zoonotic pathogens and allow for spillover. Of particular interest is data that demonstrate type I interferons (IFN), a first line defense to invading viruses, may be constitutively expressed in bats. The constant expression of type I IFNs would hamper viral infection as soon as viral invasion occurred, thereby limiting viral spread and disease. Another immunophysiological trait that may facilitate the ability to harbor viruses is a lack of somatic hypermutation and affinity maturation, which would decrease antibody affinity and neutralizing antibody titers, possibly facilitating viral persistence. -

Discovery of a Novel Human Pegivirus in Blood Associated with Hepatitis C Virus Co-Infection
RESEARCH ARTICLE Discovery of a Novel Human Pegivirus in Blood Associated with Hepatitis C Virus Co- Infection Michael G. Berg1☯, Deanna Lee2,3☯, Kelly Coller1, Matthew Frankel1, Andrew Aronsohn4, Kevin Cheng1, Kenn Forberg1, Marilee Marcinkus1, Samia N. Naccache2,3, George Dawson1, Catherine Brennan1, Donald M. Jensen4, John Hackett, Jr.1, Charles Y. Chiu2,3,5* 1 Abbott Laboratories, Abbott Park, Illinois, United States of America, 2 Department of Laboratory Medicine, University of California, San Francisco, California, United States of America, 3 UCSF-Abbott Viral Diagnostics and Discovery Center, San Francisco, California, United States of America, 4 Center for Liver Diseases, University of Chicago Medical Center, Chicago, Illinois, United States of America, 5 Department of Medicine, Division of Infectious Diseases, University of California, San Francisco, California, United States of America ☯ These authors contributed equally to this work. * [email protected] OPEN ACCESS Citation: Berg MG, Lee D, Coller K, Frankel M, Abstract Aronsohn A, Cheng K, et al. (2015) Discovery of a Novel Human Pegivirus in Blood Associated with Hepatitis C virus (HCV) and human pegivirus (HPgV), formerly GBV-C, are the only known Hepatitis C Virus Co-Infection. PLoS Pathog 11(12): human viruses in the Hepacivirus and Pegivirus genera, respectively, of the family Flaviviri- e1005325. doi:10.1371/journal.ppat.1005325 dae. We present the discovery of a second pegivirus, provisionally designated human pegi- Editor: Christian Drosten, University of Bonn, virus 2 (HPgV-2), by next-generation sequencing of plasma from an HCV-infected patient GERMANY with multiple bloodborne exposures who died from sepsis of unknown etiology. HPgV-2 is Received: September 20, 2015 highly divergent, situated on a deep phylogenetic branch in a clade that includes rodent and Accepted: November 12, 2015 bat pegiviruses, with which it shares <32% amino acid identity. -

The Breadth of Viruses in Human Semen
Article DOI: https://doi.org/10.3201/eid2311.171049 The Breadth of Viruses in Human Semen Technical Appendix Technical Appendix Table. Viruses that are capable of causing viremia and found in human semen* Detection in semen, Isolation from semen Evidence for sexual maximum detection maximum detection transmission within same Virus Family time, d time, d cohort Adenoviruses Adenoviridae AD (1) RCC Unknown Transfusion transmitted virus Anelloviridae NAA (2) No data found Unknown Lassa fever virus† Arenaviridae NAA, 103 (3) RCC, 20 (3) Unknown Rift Valley fever virus† Bunyaviridae NAA, 117 (4) No data found Unknown Ebola virus Filoviridae NAA, 531 (5) RCC, 82 (6) Epi + mol + sem (7) Marburg virus† Filoviridae AD, 83 (8) RAS, 83 (8) Epi + sem (9) GB virus C Flaviviridae NAA (10) No data found Epi + mol (11) Hepatitis C virus Flaviviridae NAA (12); AD (13) No data found Epi + mol (14) Zika virus Flaviviridae NAA, 188 (15) RCC, 7 (16) Epi + mol + sem (17) Hepatitis B virus Hepadnaviridae NAA (18); AD (19) RAS (20) Epi + mol (21) Cytomegalovirus Herpesviridae NAA (22) RCC (23) Epi + mol + sem (24) Epstein Barr virus Herpesviridae NAA (22) No data found Epi and semen (25) Human herpes virus 8 Herpesviridae NAA (26) RCC (27) Epi + mol (28) Human herpes virus 7 Herpesviridae NAA (29) No data found Unknown Human herpes virus 6 Herpesviridae NAA (22) No data found Unknown Human simplex viruses 1 and 2 Herpesviridae NAA (22); AD (1) RCC (1) Epi + mol + sem (30) Varicella zoster virus Herpesviridae NAA (22) No data found Unknown Mumps virus† Paramyxoviridae