Accurate, Fast and Lightweight Clustering of De Novo
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

The Principled Design of Large-Scale Recursive Neural Network Architectures–DAG-Rnns and the Protein Structure Prediction Problem
Journal of Machine Learning Research 4 (2003) 575-602 Submitted 2/02; Revised 4/03; Published 9/03 The Principled Design of Large-Scale Recursive Neural Network Architectures–DAG-RNNs and the Protein Structure Prediction Problem Pierre Baldi [email protected] Gianluca Pollastri [email protected] School of Information and Computer Science Institute for Genomics and Bioinformatics University of California, Irvine Irvine, CA 92697-3425, USA Editor: Michael I. Jordan Abstract We describe a general methodology for the design of large-scale recursive neural network architec- tures (DAG-RNNs) which comprises three fundamental steps: (1) representation of a given domain using suitable directed acyclic graphs (DAGs) to connect visible and hidden node variables; (2) parameterization of the relationship between each variable and its parent variables by feedforward neural networks; and (3) application of weight-sharing within appropriate subsets of DAG connec- tions to capture stationarity and control model complexity. Here we use these principles to derive several specific classes of DAG-RNN architectures based on lattices, trees, and other structured graphs. These architectures can process a wide range of data structures with variable sizes and dimensions. While the overall resulting models remain probabilistic, the internal deterministic dy- namics allows efficient propagation of information, as well as training by gradient descent, in order to tackle large-scale problems. These methods are used here to derive state-of-the-art predictors for protein structural features such as secondary structure (1D) and both fine- and coarse-grained contact maps (2D). Extensions, relationships to graphical models, and implications for the design of neural architectures are briefly discussed. -

Methodology for Predicting Semantic Annotations of Protein Sequences by Feature Extraction Derived of Statistical Contact Potentials and Continuous Wavelet Transform
Universidad Nacional de Colombia Sede Manizales Master’s Thesis Methodology for predicting semantic annotations of protein sequences by feature extraction derived of statistical contact potentials and continuous wavelet transform Author: Supervisor: Gustavo Alonso Arango Dr. Cesar German Argoty Castellanos Dominguez A thesis submitted in fulfillment of the requirements for the degree of Master’s on Engineering - Industrial Automation in the Department of Electronic, Electric Engineering and Computation Signal Processing and Recognition Group June 2014 Universidad Nacional de Colombia Sede Manizales Tesis de Maestr´ıa Metodolog´ıapara predecir la anotaci´on sem´antica de prote´ınaspor medio de extracci´on de caracter´ısticas derivadas de potenciales de contacto y transformada wavelet continua Autor: Tutor: Gustavo Alonso Arango Dr. Cesar German Argoty Castellanos Dominguez Tesis presentada en cumplimiento a los requerimientos necesarios para obtener el grado de Maestr´ıaen Ingenier´ıaen Automatizaci´onIndustrial en el Departamento de Ingenier´ıaEl´ectrica,Electr´onicay Computaci´on Grupo de Procesamiento Digital de Senales Enero 2014 UNIVERSIDAD NACIONAL DE COLOMBIA Abstract Faculty of Engineering and Architecture Department of Electronic, Electric Engineering and Computation Master’s on Engineering - Industrial Automation Methodology for predicting semantic annotations of protein sequences by feature extraction derived of statistical contact potentials and continuous wavelet transform by Gustavo Alonso Arango Argoty In this thesis, a method to predict semantic annotations of the proteins from its primary structure is proposed. The main contribution of this thesis lies in the implementation of a novel protein feature representation, which makes use of the pairwise statistical contact potentials describing the protein interactions and geometry at the atomic level. -

BIOGRAPHICAL SKETCH NAME: Berger
BIOGRAPHICAL SKETCH NAME: Berger, Bonnie eRA COMMONS USER NAME (credential, e.g., agency login): BABERGER POSITION TITLE: Simons Professor of Mathematics and Professor of Electrical Engineering and Computer Science EDUCATION/TRAINING (Begin with baccalaureate or other initial professional education, such as nursing, include postdoctoral training and residency training if applicable. Add/delete rows as necessary.) EDUCATION/TRAINING DEGREE Completion (if Date FIELD OF STUDY INSTITUTION AND LOCATION applicable) MM/YYYY Brandeis University, Waltham, MA AB 06/1983 Computer Science Massachusetts Institute of Technology SM 01/1986 Computer Science Massachusetts Institute of Technology Ph.D. 06/1990 Computer Science Massachusetts Institute of Technology Postdoc 06/1992 Applied Mathematics A. Personal Statement Advances in modern biology revolve around automated data collection and sharing of the large resulting datasets. I am considered a pioneer in the area of bringing computer algorithms to the study of biological data, and a founder in this community that I have witnessed grow so profoundly over the last 26 years. I have made major contributions to many areas of computational biology and biomedicine, largely, though not exclusively through algorithmic innovations, as demonstrated by nearly twenty thousand citations to my scientific papers and widely-used software. In recognition of my success, I have just been elected to the National Academy of Sciences and in 2019 received the ISCB Senior Scientist Award, the pinnacle award in computational biology. My research group works on diverse challenges, including Computational Genomics, High-throughput Technology Analysis and Design, Biological Networks, Structural Bioinformatics, Population Genetics and Biomedical Privacy. I spearheaded research on analyzing large and complex biological data sets through topological and machine learning approaches; e.g. -

Deep Learning in Chemoinformatics Using Tensor Flow
UC Irvine UC Irvine Electronic Theses and Dissertations Title Deep Learning in Chemoinformatics using Tensor Flow Permalink https://escholarship.org/uc/item/963505w5 Author Jain, Akshay Publication Date 2017 Peer reviewed|Thesis/dissertation eScholarship.org Powered by the California Digital Library University of California UNIVERSITY OF CALIFORNIA, IRVINE Deep Learning in Chemoinformatics using Tensor Flow THESIS submitted in partial satisfaction of the requirements for the degree of MASTER OF SCIENCE in Computer Science by Akshay Jain Thesis Committee: Professor Pierre Baldi, Chair Professor Cristina Videira Lopes Professor Eric Mjolsness 2017 c 2017 Akshay Jain DEDICATION To my family and friends. ii TABLE OF CONTENTS Page LIST OF FIGURES v LIST OF TABLES vi ACKNOWLEDGMENTS vii ABSTRACT OF THE THESIS viii 1 Introduction 1 1.1 QSAR Prediction Methods . .2 1.2 Deep Learning . .4 2 Artificial Neural Networks(ANN) 5 2.1 Artificial Neuron . .5 2.2 Activation Function . .7 2.3 Loss function . .8 2.4 Optimization . .8 3 Deep Recursive Architectures 10 3.1 Recurrent Neural Networks (RNN) . 10 3.2 Recursive Neural Networks . 11 3.3 Directed Acyclic Graph Recursive Neural Networks (DAG-RNN) . 11 4 UG-RNN for small molecules 14 4.1 DAG Generation . 16 4.2 Local Information Vector . 16 4.3 Contextual Vectors . 17 4.4 Activity Prediction . 17 4.5 UG-RNN With Contracted Rings (UG-RNN-CR) . 18 4.6 Example: UG-RNN Model of Propionic Acid . 20 5 Implementation 24 6 Data & Results 26 6.1 Aqueous Solubility Prediction . 26 6.2 Melting Point Prediction . 28 iii 7 Conclusions 30 Bibliography 32 A Source Code 37 A.1 UGRNN . -

Course Outline
Department of Computer Science and Software Engineering COMP 6811 Bioinformatics Algorithms (Reading Course) Fall 2019 Section AA Instructor: Gregory Butler Curriculum Description COMP 6811 Bioinformatics Algorithms (4 credits) The principal objectives of the course are to cover the major algorithms used in bioinformatics; sequence alignment, multiple sequence alignment, phylogeny; classi- fying patterns in sequences; secondary structure prediction; 3D structure prediction; analysis of gene expression data. This includes dynamic programming, machine learning, simulated annealing, and clustering algorithms. Algorithmic principles will be emphasized. A project is required. Outline of Topics The course will focus on algorithms for protein sequence analysis. It will not cover genome assembly, genome mapping, or gene recognition. • Background in Biology and Genomics • Sequence Alignment: Pairwise and Multiple • Representation of Protein Amino Acid Composition • Profile Hidden Markov Models • Specificity Determining Sites • Curation, Annotation, and Ontologies • Machine Learning: Secondary Structure, Signals, Subcellular Location • Protein Families, Phylogenomics, and Orthologous Groups • Profile-Based Alignments • Algorithms Based on k-mers 1 Texts | in Library D. Higgins and W. Taylor (editors). Bioinformatics: Sequence, Structure and Databanks, Oxford University Press, 2000. A. D. Baxevanis and B. F. F. Ouelette. Bioinformatics: A Practical Guide to the Analysis of Genes and Proteins, Wiley, 1998. Richard Durbin, Sean R. Eddy, Anders Krogh, -

Lior Pachter Genome Informatics 2013 Keynote
Stories from the Supplement Lior Pachter! Department of Mathematics and Molecular & Cell Biology! UC Berkeley! ! November 1, 2013! Genome Informatics, CSHL The Cufflinks supplements • C. Trapnell, B.A. Williams, G. Pertea, A. Mortazavi, G. Kwan, M.J. van Baren, S.L. Salzberg, B.J. Wold and L. Pachter, Transcript assembly and quantification by RNA-Seq reveals unannotated transcripts and isoform switching during cell differentiation, Nature Biotechnology 28, (2010), p 511–515. Supplementary Material: 42 pages. • C. Trapnell, D.G. Hendrickson, M. Sauvageau, L. Goff, J.L. Rinn and L. Pachter, Differential analysis of gene regulation at transcript resolution with RNA-seq, Nature Biotechnology, 31 (2012), p 46–53. Supplementary Material: 70 pages. A supplementary arithmetic progression? • C. Trapnell, B.A. Williams, G. Pertea, A. Mortazavi, G. Kwan, M.J. van Baren, S.L. Salzberg, B.J. Wold and L. Pachter, Transcript assembly and quantification by RNA-Seq reveals unannotated transcripts and isoform switching during cell differentiation, Nature Biotechnology 28, (2010), p 511–515. Supplementary Material: 42 pages. • C. Trapnell, D.G. Hendrickson, M. Sauvageau, L. Goff, J.L. Rinn and L. Pachter, Differential analysis of gene regulation at transcript resolution with RNA- seq, Nature Biotechnology, 31 (2012), p 46–53. Supplementary Material: 70 pages. • A. Roberts and L. Pachter, Streaming algorithms for fragment assignment, Nature Methods 10 (2013), p 71—73. Supplementary Material: 98 pages? The nature methods manuscript checklist The nature methods manuscript checklist Emperor Joseph II: My dear young man, don't take it too hard. Your work is ingenious. It's quality work. And there are simply too many notes, that's all. -

Deep Learning in Science Pierre Baldi Frontmatter More Information
Cambridge University Press 978-1-108-84535-9 — Deep Learning in Science Pierre Baldi Frontmatter More Information Deep Learning in Science This is the first rigorous, self-contained treatment of the theory of deep learning. Starting with the foundations of the theory and building it up, this is essential reading for any scientists, instructors, and students interested in artificial intelligence and deep learning. It provides guidance on how to think about scientific questions, and leads readers through the history of the field and its fundamental connections to neuro- science. The author discusses many applications to beautiful problems in the natural sciences, in physics, chemistry, and biomedicine. Examples include the search for exotic particles and dark matter in experimental physics, the prediction of molecular properties and reaction outcomes in chemistry, and the prediction of protein structures and the diagnostic analysis of biomedical images in the natural sciences. The text is accompanied by a full set of exercises at different difficulty levels and encourages out-of-the-box thinking. Pierre Baldi is Distinguished Professor of Computer Science at University of California, Irvine. His main research interest is understanding intelligence in brains and machines. He has made seminal contributions to the theory of deep learning and its applications to the natural sciences, and has written four other books. © in this web service Cambridge University Press www.cambridge.org Cambridge University Press 978-1-108-84535-9 — Deep Learning in -

Modeling and Analysis of RNA-Seq Data: a Review from a Statistical Perspective
Modeling and analysis of RNA-seq data: a review from a statistical perspective Wei Vivian Li 1 and Jingyi Jessica Li 1;2;∗ Abstract Background: Since the invention of next-generation RNA sequencing (RNA-seq) technolo- gies, they have become a powerful tool to study the presence and quantity of RNA molecules in biological samples and have revolutionized transcriptomic studies. The analysis of RNA-seq data at four different levels (samples, genes, transcripts, and exons) involve multiple statistical and computational questions, some of which remain challenging up to date. Results: We review RNA-seq analysis tools at the sample, gene, transcript, and exon levels from a statistical perspective. We also highlight the biological and statistical questions of most practical considerations. Conclusion: The development of statistical and computational methods for analyzing RNA- seq data has made significant advances in the past decade. However, methods developed to answer the same biological question often rely on diverse statical models and exhibit dif- ferent performance under different scenarios. This review discusses and compares multiple commonly used statistical models regarding their assumptions, in the hope of helping users select appropriate methods as needed, as well as assisting developers for future method development. 1 Introduction RNA sequencing (RNA-seq) uses the next generation sequencing (NGS) technologies to reveal arXiv:1804.06050v3 [q-bio.GN] 1 May 2018 the presence and quantity of RNA molecules in biological samples. Since its invention, RNA- seq has revolutionized transcriptome analysis in biological research. RNA-seq does not require any prior knowledge on RNA sequences, and its high-throughput manner allows for genome-wide profiling of transcriptome landscapes [1,2]. -

Human and Mouse Gene Structure: Comparative Analysis and Application to Exon Prediction
Downloaded from genome.cshlp.org on October 4, 2021 - Published by Cold Spring Harbor Laboratory Press Letter Human and Mouse Gene Structure: Comparative Analysis and Application to Exon Prediction Serafim Batzoglou,1,4,7 Lior Pachter,2,7 Jill P. Mesirov,3 Bonnie Berger,1,4,6 and Eric S. Lander3,5,6 1Laboratory for Computer Science, Massachusetts Institute of Technology, Cambridge, Massachusetts 02139 USA; 2Department of Mathematics, University of California Berkeley, Berkeley, California 94720 USA; 3Whitehead Institute for Biomedical Research, Cambridge, Massachusetts 02142 USA; 4Department of Mathematics, Massachusetts Institute of Technology, Cambridge, Massachusetts 02139 USA; 5Department of Biology, Massachusetts Institute of Technology, Cambridge, Massachusetts 02139 USA We describe a novel analytical approach to gene recognition based on cross-species comparison. We first undertook a comparison of orthologous genomic loci from human and mouse, studying the extent of similarity in the number, size and sequence of exons and introns. We then developed an approach for recognizing genes within such orthologous regions by first aligning the regions using an iterative global alignment system and then identifying genes based on conservation of exonic features at aligned positions in both species. The alignment and gene recognition are performed by new programs called GLASS and ROSETTA, respectively. ROSETTA performed well at exact identification of coding exons in 117 orthologous pairs tested. A fundamental task in analyzing genomes is to identify by comparison of syntenic human and mouse genomic the genes. This is relatively straightforward for organ- sequences. isms with compact genomes (such as bacteria, yeast, It is well known that cross-species sequence com- flies and worms) because exons tend to be large and the parison can help highlight important functional ele- introns are either non-existent or tend to be short. -

Eric S. Lander
Chancellor's Distinguished Fellows Program 2004-2005 Selective Bibliography UC Irvine Libraries Eric S. Lander February 25, 2005 Prepared by: John E. Sisson Biological Sciences Librarian [email protected] Journal Articles (Selected from over 220 articles he has authored or co-authored) Poirier, C., Y. J. Qin, C.P. Adams, Y. Anaya, J. B. Singer, A. E. Hill, Eric S. Lander, J.H. Nadeau, and C. E. Bishop. "A complex interaction of imprinted and maternal-effect genes modifies sex determination in odd sex (Ods) mice." Genetics 168, no. 3 (2004): 1557-1562. Skuse, D. H., S. Purcell, M. J. Daly, R. J. Dolan, J. S. Morris, K. Lawrence, Eric S. Lander, and P. Sklar. "What can studies on Turner syndrome tell us about the role of X- linked genes in social cognition?." American Journal of Medical Genetics Part B- Neuropsychiatric Genetics 130B, no. 1 (2004): 8-9. Rioux, J. D., H. Karinen, K. Kocher, S. G. McMahon, P. Karkkainen, E. Janatuinen, M. Heikkinen, R. Julkunen, J. Pihlajamaki, A. Naukkarinen, V. M. Kosma, M. J. Daly, Eric S. Lander, and M. Laakso. "Genomewide search and association studies in a Finnish celiac disease population: Identification of a novel locus and replication of the HLA and CTLA4 loci." American Journal of Medical Genetics Part A 130A, no. 4 (2004): 345- 350. Michalkiewicz, M., T. Michalkiewicz, R. A. Ettinger, E. A. Rutledge, J. M. Fuller, D. H. Moralejo, B. Van Yserloo, A. J. MacMurray, A. E. Kwitek, H. J. Jacob, Eric S. Lander, and A. Lernmark. "Transgenic rescue demonstrates involvement of the Ian5 gene in T cell development in the rat." Physiological Genomics 19, no. -

Bringing Folding Pathways Into Strand Pairing Prediction
Bringing folding pathways into strand pairing prediction Jieun Jeong1,2, Piotr Berman1, and Teresa Przytycka2 1 Computer Science and Engineering Department The Pennsylvania State University University Park, PA 16802 USA 2 National Center for Biotechnology Information US National Library of Medicine, National Institutes of Health Bethesda, MD 20894 email: [email protected], [email protected], [email protected] Abstract. The topology of β-sheets is defined by the pattern of hydrogen- bonded strand pairing. Therefore, predicting hydrogen bonded strand partners is a fundamental step towards predicting β-sheet topology. In this work we report a new strand pairing algorithm. Our algorithm at- tempts to mimic elements of the folding process. Namely, in addition to ensuring that the predicted hydrogen bonded strand pairs satisfy basic global consistency constraints, it takes into account hypothetical folding pathways. Consistently with this view, introducing hydrogen bonds be- tween a pair of strands changes the probabilities of forming other strand pairs. We demonstrate that this approach provides an improvement over previously proposed algorithms. 1 Introduction The prediction of protein structure from protein sequence is a long-held goal that would provide invaluable information regarding the function of individ- ual proteins and the evolution of protein families. The increasing amount of sequence and structure data, made it possible to decouple the structure predic- tion problem from the problem of modeling of protein folding process. Indeed, a significant progress has been achieved by bioinformatics approaches such as homology modeling, threading, and assembly from fragments [16]. At the same time, the fundamental problem of how actually a protein acquires its final folded state remains a subject of controversy. -
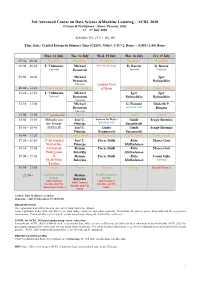
ACDL-2020-Programme-Ver10.0
3rd Advanced Course on Data Science &Machine Learning – ACDL 2020 Certosa di Pontignano - Siena, Tuscany, Italy 13 – 17 July 2020 Schedule Ver. 11.0 – July 9th Time Zone: Central European Summer Time (CEST), Offset: UTC+2, Rome – (GMT+2:00) Rome Mon, 13 July Tue, 14 July Wed, 15 July Thu, 16 July Fri, 17 July 07:30 – 09:00 Breakfast Breakfast Breakfast Breakfast Breakfast 09:00 – 09:50 T. Viehmann Michael 8:30 Social Tour D. Bacciu D. Bacciu Tutorial Bronstein Tutorial Tutorial 09:50 – 10:40 Michael Igor Bronstein Babuschkin Tutorial Guided Visit 10:40 – 11:20 Coffee break Coffee break of Siena Coffee break Coffee break 11:20 – 12:10 T. Viehmann Michael Igor Igor Tutorial Bronstein Babuschkin Babuschkin Tutorial 12:10 – 13:00 Michael G. Fiameni Diederik P. Bronstein Industrial Talk Kingma Tutorial 13:00 – 15:00 Lunch (13:00-14:00) Lunch Lunch Lunch Lunch 15:00 – 15:50 Mihaela van José C. Lorenzo De Mattei Guido Sergiy Butenko der Schaar Principe Industrial Talk Sanguinetti 15:50 – 16:40 (14:00-16:40) José C. Guido Guido Sergiy Butenko Principe Sanguinetti Sanguinetti 16:40 – 17:20 Coffee break Coffee break Coffee break Coffee break Coffee break 17:20 – 18:10 17:20 Guided José C. Pierre Baldi Risto Marco Gori Visit of the Principe Miikkulainen 18:10 – 19:00 Certosa di Roman Pierre Baldi Risto Marco Gori Pontignano Belavkin Miikkulainen 19:00 – 19:50 & Roman Pierre Baldi Risto Varun Ojha 18:20 Wine Belavkin Miikkulainen Tutorial Tasting 19:50 – 21:50 Dinner Dinner Dinner Dinner Social Dinner 21:50 – Oral Presentation Roman Oral Presentation Session Belavkin Session with Cantucci with Cantucci with Cantucci biscuits and Vin biscuits and Vin biscuits and Vin Santo (sweet wine) Santo Santo Arrival: July 12 (Dinner at 20:30) Departure: July 18 (Breakfast 07:30-09:00) REGISTRATION The registration desk will be located close to the Main Conference Room.