Huisgen and His Adventures in a Playground of Mechanisms and Novel Reactions
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Program & Abstracts
Program & Abstracts Conference venue: University of Łódź, Faculty of Chemistry, Tamka-Str. 12, The Large Hall, A-1 Organizing committee: Chairman: Prof. dr. hab. Grzegorz Mlostoń Secretary: Prof. UŁ, dr. hab. Jarosław Romański Members: Dr. Katarzyna Urbaniak Dr. Marcin Jasiński Małgorzata Celeda Zenona Frydrych Organizing Committee thanks to WITKO company and TriMen Chemicals for support during Mini-Symposium VIIIth International Mini-Symposium ‘'The Huisgen Cycloaddition Reaction as a Universal Tool in Chemistry, Biology, and Medicine' Program Special Guest Lecture 11:30-12:15 Wolfgang Weigand University of Jena, Germany 12:15-13:15 Lunch 13:25 Official Opening Session 1: Chairman: Prof. Zbigniew Kamioski Keynote Lecture 13:30 – 14:15 Hans Ulrich Reissig KL Free University of Berlin, Germany Invited Lectures 14:15 – 14:45 Karol Kacprzak IL-1 Adam Mickiewicz University in Poznao, Poland 14:45 – 15:15 Wafaa Abdou IL-2 National Research Center in Cairo, Egypt 15:15 – 15:45 Zbigniew Leśnikowski IL-3 Institute of Medicinal Biology, PAS Łódź, Poland 15:45 – 16:15 Coffee break Session 2 Chairman: Prof. Piotr Kiełbasioski Invited Lectures 16:15 – 16:45 Peter R. Schreiner IL-4 Justus Liebig University in Giessen, Germany 16:45 – 17:15 Keith ó Proinsiasa IL-5 Institute of Organic Chemistry, PAS Warsaw, Poland 17:15 – 17:45 Florence Dumarcay-Charbonnier IL-6 University of Lorraine, Nancy, France Short Lectures 17:45 – 18:00 Marcin Jasioski SL-1 University of Łódź, Poland 18:00 – 18:15 Bogna Rudolf SL-2 University of Łódź, Poland 18:15 – …… Garden -

UCLA Electronic Theses and Dissertations
UCLA UCLA Electronic Theses and Dissertations Title Computational Studies on the Reactivity, Selectivity and Molecular Dynamics of Cycloaddition Reactions Permalink https://escholarship.org/uc/item/1jk894mq Author Yu, Peiyuan Publication Date 2017 Peer reviewed|Thesis/dissertation eScholarship.org Powered by the California Digital Library University of California UNIVERSITY OF CALIFORNIA Los Angeles Computational Studies on the Reactivity, Selectivity and Molecular Dynamics of Cycloaddition Reactions A dissertation submitted in partial satisfaction of the requirements for the degree Doctor of Philosophy in Chemistry by Peiyuan Yu 2017 © Copyright by Peiyuan Yu 2017 ABSTRACT OF THE DISSERTATION Computational Studies on the Reactivity, Selectivity and Molecular Dynamics of Cycloaddition Reactions by Peiyuan Yu Doctor of Philosophy in Chemistry University of California, Los Angeles, 2017 Professor Kendall N. Houk, Chair The first part of this dissertation describes computational studies of higher-order cycloadditions with a focus on elucidating the origins of periselectivity. These reactions involve ambimodal transition states (TS) and bifurcations of potential energy surface. Chapter 1 describes density functional theory (DFT) studies of a transannular [6+4] cycloaddition proposed in the biosynthesis of heronamide A, in which an unbridged 10- membered ring is formed. Ring and steric strains are found to be essential in controlling the product stability that makes this reaction feasible. Chapter 2 presents an Environment-Perturbed Transition State Sampling method to explore the mechanism of the enzyme SpnF-catalyzed Diels–Alder. A [6+4] cycloaddition is also involved and enzyme enhances the formation of [4+2] product, with respect to the counterparts in the gas phase and in water. Chapter 3 explores the mechanisms and selectivities of the cycloadditions of tropone to dimethylfulvene discovered ii in 1967 by Houk. -

Recent Advances on Mechanistic Studies on C–H Activation
Open Chem., 2018; 16: 1001–1058 Review Article Open Access Daniel Gallego*, Edwin A. Baquero Recent Advances on Mechanistic Studies on C–H Activation Catalyzed by Base Metals https:// doi.org/10.1515/chem-2018-0102 received March 26, 2018; accepted June 3, 2018. 1Introduction Abstract: During the last ten years, base metals have Application in organic synthesis of transition metal- become very attractive to the organometallic and catalytic catalyzed cross coupling reactions has been positioned community on activation of C-H bonds for their catalytic as one of the most important breakthroughs during the functionalization. In contrast to the statement that new millennia. The seminal works based on Pd–catalysts base metals differ on their mode of action most of the in the 70’s by Heck, Noyori and Suzuki set a new frontier manuscripts mistakenly rely on well-studied mechanisms between homogeneous catalysis and synthetic organic for precious metals while proposing plausible chemistry [1-5]. Late transition metals, mostly the precious mechanisms. Consequently, few literature examples metals, stand as the most versatile catalytic systems for a are found where a thorough mechanistic investigation variety of functionalization reactions demonstrating their have been conducted with strong support either by robustness in several applications in organic synthesis [6- theoretical calculations or experimentation. Therefore, 12]. Owing to the common interest in the catalysts mode we consider of highly scientific interest reviewing the of action by many research groups, nowadays we have a last advances on mechanistic studies on Fe, Co and Mn wide understanding of the mechanistic aspects of precious on C-H functionalization in order to get a deep insight on metal-catalyzed reactions. -

11 Concerted Pericyclic Reactions
11 Concerted Pericyclic Reactions Introduction There are many reactions in organic chemistry that give no evidence of involving intermediates when subjected to the usual probes for studying reaction mechanisms. Highly polar transition states do not seem to be involved either,since the rates of the reactions are often insensitive to solvent polarity. Efforts to detect free-radical intermedi- ates in these reactions by spectroscopic or chemical means have not been successful,and the reaction rates are neither increased by initiators nor decreased by inhibitors of free- radical reactions. This absence of evidence of intermediates leads to the conclusion that the reactions are single-step processes in which bond making and bond breaking both contribute to the structure at the transition state,although not necessarily to the same degree. Such processes are called concerted reactions. There are numerous examples of both unimolecular and bimolecular concerted reactions. An important group of concerted reactions are the concerted pericyclic reactions.1 A pericyclic reaction is characterized as a change in bonding relationship that takes place as a continuous concerted reorganization of electrons. The word ``concerted'' speci®es that there is a single transition state and that there are no intermediates in the process. To maintain continuous electron ¯ow,pericyclic reactions must occur through cyclic transi- tion states. Furthermore,the cyclic transition state must correspond to an arrangement of the participating orbitals that can maintain a bonding interaction between the reacting atoms throughout the course of the reaction. We shall see shortly that these requirements make pericyclic reaction highly predictable in terms of such features as relative reactivity, stereospeci®city,and regioselectivity. -

2.1 the 18-ELECTRON RULE the 18E Rule1 Is a Way to Help Us Decide
30 GENERAL PROPERTIES OF ORGANOMETALLIC COMPLEXES 2.1 THE 18-ELECTRON RULE The 18e rule1 is a way to help us decide whether a given d-block transition metal organometallic complex is likely to be stable. Not all the organic formulas we can write down correspond to stable species. For example, CH5 requires a 5-valent carbon and is therefore not stable. Stable compounds, such as CH4,have the noble gas octet, and so carbon can be thought of as following an 8e rule. This corresponds to carbon using its s and three p orbitals to form four filled bonding orbitals and four unfilled antibonding orbitals. On the covalent model, we can consider that of the eight electrons required to fill the bonding orbitals, four come from carbon and one each comes from the four H substituents. We can therefore think of each H atom as being a 1e ligand to carbon. To assign a formal oxidation state to carbon in an organic molecule, we impose an ionic model by artificially dissecting it into ions. Each electron pair in any bond is assigned to the most electronegative of the two atoms or groups that constitute the bond. For methane, this dissection gives C4− + 4H+, with carbon as the more electronegative element. This makes methane an 8e compound with an oxidation state of −4, usually written C(-IV). Note that the net electron count always remains the same, whether we adopt the covalent (4e {Catom}+4 × 1e {4H atoms}=8e) or ionic (8e{C4−ion}+4 × 0e{4H+ions}=8e) model. -

Assessing the Feasibility of a One-Pot, Tandem Olefin Metathesis and Isomerization Sequence to Synthesize Conjugated Aromatic Olefins
University of Tennessee at Chattanooga UTC Scholar Student Research, Creative Works, and Honors Theses Publications 8-2016 Assessing the feasibility of a one-pot, tandem olefin metathesis and isomerization sequence to synthesize conjugated aromatic olefins Ajay D. Makwana University of Tennessee at Chattanooga, [email protected] Follow this and additional works at: https://scholar.utc.edu/honors-theses Part of the Chemistry Commons Recommended Citation Makwana, Ajay D., "Assessing the feasibility of a one-pot, tandem olefin metathesis and isomerization sequence to synthesize conjugated aromatic olefins" (2016). Honors Theses. This Theses is brought to you for free and open access by the Student Research, Creative Works, and Publications at UTC Scholar. It has been accepted for inclusion in Honors Theses by an authorized administrator of UTC Scholar. For more information, please contact [email protected]. Assessing the Feasibility of a One-Pot, Tandem Olefin Metathesis and Isomerization Sequence to Synthesize Conjugated Aromatic Olefins Ajay D. Makwana Departmental Honors Thesis The University of Tennessee at Chattanooga Department of Chemistry Project Director: Dr. Kyle S. Knight Examination Date: April 4, 2016 Members of Examination Committee: Dr. John P. Lee Dr. Han J. Park Dr. David Giles 1 Abstract The synthesis of substituted phenylpropene dimers using a one-pot, tandem olefin metathesis and isomerization sequence has been studied. This sequence relies on the facilitated, in-situ conversion of a ruthenium carbene species (Ru=C) to a ruthenium hydride species (Ru-H) upon addition of an inorganic hydride source. Three separate reactions occur within one reaction flask: 1) olefin metathesis of the starting phenylpropene to yield phenylpropene dimer via Ru=C catalyst, 2) conversion of Ru=C to Ru-H via addition of an inorganic hydride source, 3) isomerization of phenylpropene dimer via insertion and β-hydride elimination to yield conjugated product. -

Wilkinson's Catalyst
Homogeneous Catalytic Processes Hydroformylation RCH2CH2 O RCH CH2 + CO + H2 Co(I), Rh(I) or Pt(II) H Oxidation H3C O H2CCH2 + O2 Pd(II) or Cu(II) H Carbonylation H3C O - CH3OH + CO [RhI2(CO)2] OH Hydrocyanation [Ni{P(OR) } ] H CCC CH + 2HCN 3 4 NCCH CH CH CH CN 2 H H 2 2 2 2 2 Cyclotrimerization Ni(acac) 3 HC CH 2 Hydrogenation of Alkenes The most commonly used catalyst is the Wilkinson’s Catalyst Many alkenes are hydrogenated with hydrogen at 1atm pressure or less . Wilkinson’s catalyst is highly sensitive to the nature of the phosphine ligand and the alkene substrate. Analogous catalysts with alkyl phosphine ligands are inactive Highly hindered alkenes and ethylene are not hydrogenated by the catalyst Wilkinson’s Catalyst Ph3P PPh3 R CH3 Rh H2 Ph3P Cl Reductive Elimination Oxidative Addition R H H PPh3 H PPh3 Rh Rh Ph3P Cl Ph3P Cl PPh3 PPh3 Ligand Dissociation Ligand Association PPh3 PPh3 H R H PPh3 Rh H PPh3 Cl Rh Ph3P Cl PPh3 H Migratory Insertion H PPh3 Rh R Ph3P Cl Alkene Coordination R Hydroformylation Catalyst + CO + H2 RCH2CH2CHO R A less common,,pppyy but more appropriate name is hydrocarbonylation Both cobalt and rhodium complexes are used as catalysts. Alkene iitiisomerization, alkene hdhydrogena tion and ftiformation of bhdbranched alde hy des are the possible side reactions. Cobalt catalysts operate at 150 ºC and 250 atm, whereas Rhodium catalysts operate at moderate temperatures and 1 atm. Rhodium catalysts promotes the formation of linear aldehydes. Cobalt catalysts do so if modified with alkylphosphine ligands. -

Automated Calculation of Reaction Kinetics Via Transition State Theory
AUTOMATED CALCULATION OF REACTION KINETICS VIA TRANSITION STATE THEORY A Dissertation Presented By Pierre Lennox Bhoorasingh to The Department of Chemical Engineering In partial fulfillment of the requirements for the degree of Doctor of Philosophy in the field of Chemical Engineering Northeastern University Boston, Massachusetts August 2016 Dedication I dedicate this thesis to AMT. i Acknowledgments I have been able to complete this thesis work due to the help I have received from those who have found time in their busy schedules. This is my attempt to express my profound gratitude to those who have helped me during my thesis work. Thanks to my advisor, Prof. Richard West, for the guidance over the 5 years. You also gave me the freedom to explore and that has only enhanced my thesis work, and it has been a pleasure to be your first graduate student. I would also like to thank my thesis committee members, Dr. David Budil, Dr. Hicham Fenniri, Dr. C. Franklin Goldsmith, and Dr. Reza Sheikhi. They made the time to have engaging discussions that impacted this thesis, and were also very generous with their professional advice. Thanks to the Computational Modeling group. Fariba Seyedzadeh Khanshan and Be- linda Slakman, you were always helpful in our discussions and made the laboratory a fun working environment. I’d also like to thank Jason Cain for being a super helpful under- graduate who assumed nothing in pursuit of the right approach. I want to also thank Sean Troiano, Victor Lambert, Jacob Barlow, and Elliot Nash for their contributions to laboratory discussions. -
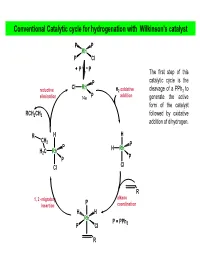
Conventional Catalytic Cycle for Hydrogenation with Wilkinson's Catalyst
Conventional Catalytic cycle for hydrogenation with Wilkinson’s catalyst P P Rh P Cl PP The first step of this P catalytic cycle is the Cl Rh reductive H2 oxidative cleavage of a PPh3 to elimination 14e P addition generate the active form of the catalyst RCH2CH3 followed by oxidative addition of dihydrogen. R H H CH 2 P P H Rh H2C Rh P P Cl Cl R alkene 1, 2 -migratory P insertion coordination H H Rh P = PPh P Cl 3 R AJELIAS L7-S18 catalytic cycle for hydrogenation H H2 oxidative P P addition H P Kinetic studies have Rh Rh shown that the P Cl P Cl dissociation of PPh3 P from the distorted P square planar complex PP (due to trans RhCl(PPh3)3 in effect of H ) H benzene occurs only to a very small extent H2 oxidative P (k = 2.3 × 10–7 M at P addition HRh Cl Rh 25°C), and P P under an atmosphere Cl of H2, a solution of RCH2CH3 RhCl(PPh3)3 becomes reductive alkene yellow as a result of elimination the oxidative addition of H2 to give cis- R H P CH2 H2RhCl(PPh3)3. P H H H2CRh Rh P 1, 2 -migratory P Cl insertion Cl R The trans effect is the labilization (making unstable) of ligands that are trans to certain other ligands, which can thus be regarded as trans-directing ligands. The intensity of the trans effect (as measured by the increase in rate of substitution of the trans ligand) follows this sequence: H2O, OH− < NH3 < py < Cl− < Br− < I−, < PR3, CH3− < H−, NO, CO AJELIAS L7-S19 Relative reactivity of alkenes for homogenous catalytic hydrogenation • Cis alkenes undergo hydrogenation more readily than trans alkenes •Internal and branched alkenes -

Pericyclic Reactions Notes
- 1 - PERICYCLIC REACTIONS NOTES Pericyclic reactions cannot be treated adequately by “curly-arrow” formalisms and a knowledge of molecular orbital theory is crucial to their understanding. They are reactions in which “all first order changes in bonding relationships takes place in concert on a closed curve” (Woodward & Hoffmann). More simply, the term “pericyclic” covers all concerted reactions involving a cyclic flow of electrons through a single transition state. Pericyclic reactions can be predicted and controlled to a great degree, which makes them very useful in synthesis. There are broadly four classes of pericyclic reaction: Sigmatropic – These are unimolecular isomerisations, and involve the movement of a σ-bond from one position to another. An illustration would be the first step of the Claisen Rearrangement: Note the nomenclature of this reaction, being described as a [i,j] shift. For example, this following is a [1,7] shift: Electrocyclic – These are unimolecular. They are characterised by ring opening or closing with a σ- bond forming at one end. Ring closing is more common, since this is formation of a σ-bond at the expense of a π-bond, but ring strain can lead to opening. Two examples are: Cycloaddition – This is the largest class of pericyclic reaction. It is characterised by two fragments coming together to form two new σ-bonds in a ring. Some examples are Diels-Alder and Ozonolysis reactions, which are described below. Chelotropic reactions are a specific type of cycloaddition, where the two bonds are made or broken at the same atom. The classic example of this is carbene addition to a double bond. -

Basics of Catalysis and Kinetics
Basics of Catalysis and Kinetics Nobel laureates in catalysis: Haber (1918) Ziegler and Natta (1963) Wilkinson, Fischer (1973) Knowles, Noyori, Sharpless (2001) Grubbs, Schrock, Chauvin (2006) Ertl (2007) Heck, Negishi, Suzuki (2010) G. Poli Definition The concept of catalyst was first introduced by Berzelius in the 19th century. Subsequently, Ostwald came up with the definition that we still use today: “A catalyst is a substance that increases the rate of a chemical reaction, without being consumed or produced”… Berzelius, J. J. Ann. Chim. Phys. 1836, 61, 146-151 Ostwald, W. Z. Phys. Chem. 1894, 15, 705-706 G. Poli The Catalyst and the Rate of a Reaction ΔG non catalyzed catalyzed A B Reaction coordinate The catalyst changes only the rate of the reaction, and does not change the thermodynamics. However, the temperature can affect equilibrium concentrations: ΔG = ΔH - T ΔS G. Poli Historical basis for the Analysis of Catalytic Kinetics The Michaelis-Menten Kinetics G. Poli Thermal, Catalyzed, Inhibited, and Promoted Reactions I II III IV uncatalyzed catalysis inhibition stoichimetric process (no catalysis) promotion (no catalysis) A A A A Acat Bcat Ainhib Binhib Aprom B B (+ cat) B Bprom SM is stabilized SM is stabilized more than TS --> a further reaction is less than TS the uncatalyzed path needed to liberate B is preferred G. Poli Thermal, Catalyzed, Inhibited, and Promoted Reactions O NH HN CCl3 Ar O CCl3 xylenes 140°C type I Ar CCl3 CCl3 O NH PdCl2(PhCN)2 (5 mol%) O NH type II THF, rt N H H N B N N N N B Pt N N 1. -

Neural Networks in Chemistry
Neural Networks in Chemistry By Johann Gasteiger” and Jure Zupan” The capabilities of the human brain have always fascinated scientists and led them to investi- gate its inner workings. Over the past 50 years a number of models have been developed which have attempted to replicate the brain’s various functions. At the same time the development of computers was taking a totally different direction. As a result, today’s computer architec- tures, operating systems, and programming have very little in common with information processing as performed by the brain. Currently we are experiencing a reevaluation of the brain’s abilities, and models of information processing in the brain have been translated into algorithms and made widely available. The basic building-block of these brain models (neural networks) is an information processing unit that is a model of a neuron. An artificial neuron of this kind performs only rather simple mathematical operations; its effectiveness is derived solely from the way in which large numbers of neurons may be connected to form a network. Just as the various neural models replicate different abilities of the brain, they can be used to solve different types of problem: the classification of objects, the modeling of functional relationships, the storage and retrieval of information, and the representation of large amounts of data. This potential suggests many possibilities for the processing of chemical data, and already applications cover a wide area: spectroscopic analysis, prediction of reactions, chem- ical process control, and the analysis of electrostatic potentials. All these are just a small sample of the great many possibilities.