Patients with Noonan Syndrome Phenotype: Spectrum of Clinical Features and Congenital Heart Defect S
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-
Basic Concepts in Basic Concepts in Dysmorphology
Basic Concepts in Dysmorphology Samia Temtamy* & Mona Aglan** *Professor of Human Genetics **Professor of Clinical Genetics Human Genetics & Genome Research Division National Research Centre, Cairo, Egypt OtliOutline y Definition of dysmorphology y Definition of terms routinely used in the description of birth defects y Impact of malformations y The difference between major & minor anomalies y Approach to a dysmorphic individual: y Suspicion & analysis y Systematic physical examination y CfitifdiConfirmation of diagnos is y Intervention y Summary 2 DfiitiDefinition of fd dysmorph hlology y The term “dysmorphology” was first coined by Dr. DidSithUSAiDavid Smith, USA in 1960s. y It implies study of human congenital defects and abnormalities of body structure that originate before birth. y The term “dysmorphic” is used to describe individuals whose physical fffeatures are not usually found in other individuals with the same age or ethnic background. y “Dys” (Greek)=disordered or abnormal and “Morph”=shape 3 Definition of terms routinely used in the d escri pti on of bi rth d ef ect s y A malformation / anomaly: is a primary defect where there i s a bas ic a ltera tion o f s truc ture, usuall y occurring before 10 weeks of gestation. y Examples: cleft palate, anencephaly, agenesis of limb or part of a limb. 4 Cleft lip & palate Absence of digits (ectrodactyly) y Malformation Sequence: A pattern of multiple defects resulting from a single primary malformation. y For example: talipes and hydrocephalus can result from a lumbar neural tube defect. Lumbar myelomeningeocele 5 y Malformation Syndrome: A pattern of features, often with an underlying cause, that arises from several different errors in morphogenesis. -

Dysmorphology Dysmorphism
Dysmorphology Carolyn Jones, M.D., PhD Dysmorphism Morphologic developmental abnormalities. This may been seen in many syndromes of genetic or environmental origin. 1 Malformation A recognized dysmorphic feature. A structure not formed correctly. This can either be the cause of genetic factors or environment. 2 Deformation An external force resulting in the inability of a structure to form correctly. Example: club feet in a woman with oligohydramnios, fibroid tumors or multiple gestation. Disruption Birth defect resulting from the destruction of a normally forming structure. This can be caused by vascular occlusion, teratogen, or rupture of amniotic sac (amniotic band syndrome). 3 Common Dysmorphic features Wide spacing between eyes, hypertelorism Narrow spacing between eyes, hypotelorism Palpebral fissure length Epicanthal folds Common Dysmorphic Features Philtrum length Upper lip Shape of nose 4 Syndrome A number of malformations seen together Cause of Syndromes Chromosomal aneuploidy Single Gene abnormalities Teratogen exposure Environmental 5 Chromosomal Aneuploidy Nondisjuction resulting in the addition or loss of an entire chromosome Deletion of a part of a chromosome Microdeletion syndromes (small piece missing which is usually only detected using special techniques) 6 Common Chromosomal Syndromes caused by Nondisjuction Down syndrome (trisomy 21) Patau syndrome (trisomy 13) Edwards syndrome (trisomy 18) Turner syndrome (monosomy X) Klinefelter syndrome (47,XXY) Clinical Features at birth of Down syndrome Low set small ears Hypotonia Simian crease Wide space between first and second toe Flat face 7 Clinical Features of Down Syndrome Small stature Congenital heart defects (50-70%) Acquired and congenital hearing impairment. Duodenal atresia Hirshprungs disease Trisomy 13 Occurs in 1/5000 live births. -

Medical Genetics and Genomic Medicine in the United States of America
View metadata, citation and similar papers at core.ac.uk brought to you by CORE provided by George Washington University: Health Sciences Research Commons (HSRC) Himmelfarb Health Sciences Library, The George Washington University Health Sciences Research Commons Pediatrics Faculty Publications Pediatrics 7-1-2017 Medical genetics and genomic medicine in the United States of America. Part 1: history, demographics, legislation, and burden of disease. Carlos R Ferreira George Washington University Debra S Regier George Washington University Donald W Hadley P Suzanne Hart Maximilian Muenke Follow this and additional works at: https://hsrc.himmelfarb.gwu.edu/smhs_peds_facpubs Part of the Genetics and Genomics Commons APA Citation Ferreira, C., Regier, D., Hadley, D., Hart, P., & Muenke, M. (2017). Medical genetics and genomic medicine in the United States of America. Part 1: history, demographics, legislation, and burden of disease.. Molecular Genetics and Genomic Medicine, 5 (4). http://dx.doi.org/10.1002/mgg3.318 This Journal Article is brought to you for free and open access by the Pediatrics at Health Sciences Research Commons. It has been accepted for inclusion in Pediatrics Faculty Publications by an authorized administrator of Health Sciences Research Commons. For more information, please contact [email protected]. GENETICS AND GENOMIC MEDICINE AROUND THE WORLD Medical genetics and genomic medicine in the United States of America. Part 1: history, demographics, legislation, and burden of disease Carlos R. Ferreira1,2 , Debra S. Regier2, Donald W. Hadley1, P. Suzanne Hart1 & Maximilian Muenke1 1National Human Genome Research Institute, National Institutes of Health, Bethesda, Maryland 2Rare Disease Institute, Children’s National Health System, Washington, District of Columbia Correspondence Carlos R. -

Medical Genetics and Genomic Medicine in the United States of America
Himmelfarb Health Sciences Library, The George Washington University Health Sciences Research Commons Pediatrics Faculty Publications Pediatrics 7-1-2017 Medical genetics and genomic medicine in the United States of America. Part 1: history, demographics, legislation, and burden of disease. Carlos R Ferreira George Washington University Debra S Regier George Washington University Donald W Hadley P Suzanne Hart Maximilian Muenke Follow this and additional works at: https://hsrc.himmelfarb.gwu.edu/smhs_peds_facpubs Part of the Genetics and Genomics Commons APA Citation Ferreira, C., Regier, D., Hadley, D., Hart, P., & Muenke, M. (2017). Medical genetics and genomic medicine in the United States of America. Part 1: history, demographics, legislation, and burden of disease.. Molecular Genetics and Genomic Medicine, 5 (4). http://dx.doi.org/10.1002/mgg3.318 This Journal Article is brought to you for free and open access by the Pediatrics at Health Sciences Research Commons. It has been accepted for inclusion in Pediatrics Faculty Publications by an authorized administrator of Health Sciences Research Commons. For more information, please contact [email protected]. GENETICS AND GENOMIC MEDICINE AROUND THE WORLD Medical genetics and genomic medicine in the United States of America. Part 1: history, demographics, legislation, and burden of disease Carlos R. Ferreira1,2 , Debra S. Regier2, Donald W. Hadley1, P. Suzanne Hart1 & Maximilian Muenke1 1National Human Genome Research Institute, National Institutes of Health, Bethesda, Maryland 2Rare Disease Institute, Children’s National Health System, Washington, District of Columbia Correspondence Carlos R. Ferreira, National Human Genome Research Institute, National Institutes of Health, 10 Center Drive, Building 10, Room 10C103, Bethesda, Maryland 20892-1851. -

A Comprehensive Examination of Human Triploidy and Diploid/Triploid Mixoploidy
A COMPREHENSIVE EXAMINATION OF HUMAN TRIPLOIDY AND DIPLOID/TRIPLOID MIXOPLOIDY by Jason Christopher Carson BS, Allegheny College, 2004 Submitted to the Graduate Faculty of the Graduate School of Public Health in partial fulfillment of the requirements for the degree of Master of Science University of Pittsburgh 2009 UNIVERSITY OF PITTSBURGH GRADUATE SCHOOL OF PUBLIC HEALTH This thesis was presented by Jason Christopher Carson It was defended on May 27, 2009 and approved by Thesis Advisor: Urvashi Surti Ph.D Associate Professor Pathology School of Medicine University of Pittsburgh Committee Member: Eleanor Feingold Ph.D. Associate Professor Human Genetics Graduate School of Public Health University of Pittsburgh Committee Member: Susanne M. Gollin Ph.D. Professor Human Genetics Graduate School of Public Health University of Pittsburgh ii Copyright © by Jason C. Carson 2009 iii Urvashi Surti, Ph.D. A COMPREHENSIVE EXAMINATION OF HUMAN TRIPLOIDY AND DIPLOID/TRIPLOID MIXOPLOIDY Jason C. Carson, MS University of Pittsburgh, 2009 Triploidy is the presence of 69 chromosomes instead of the normal diploid number of 46 and can occur in a complete form or in a mixoploid state in which there are populations of diploid and triploid cells in the same individual. The extra haploid set can be of paternal or maternal origin. Triploidy is one of the most common chromosome aberrations seen in 1-2% of all recognized pregnancies and can lead to partial mole which can in turn lead to serious complications for the mother and fetus. Given the high incidence of chromosome abnormalities including triploidy and its impact on individuals with chromosomally abnormal pregnancies, a greater understanding of their etiology has a potential to contribute greatly to public health by enhancing the management and possible future prevention. -

Face Processing in Turner Syndrome
Face Processing in Turner syndrome Kate Louise Elgar Thesis submitted for the degree of Doctor of Philosophy September 2002 Institute of Child Health, University College London ProQuest Number: 10015080 All rights reserved INFORMATION TO ALL USERS The quality of this reproduction is dependent upon the quality of the copy submitted. In the unlikely event that the author did not send a complete manuscript and there are missing pages, these will be noted. Also, if material had to be removed, a note will indicate the deletion. uest. ProQuest 10015080 Published by ProQuest LLC(2016). Copyright of the Dissertation is held by the Author. All rights reserved. This work is protected against unauthorized copying under Title 17, United States Code. Microform Edition © ProQuest LLC. ProQuest LLC 789 East Eisenhower Parkway P.O. Box 1346 Ann Arbor, Ml 48106-1346 Abstract This thesis explored the influence of X-linked genes on the development of face- processing abilities. It assessed face-processing abilities in women with Turner syndrome (TS) who have just one, instead of two, X-chromosomes. Study One assessed the nature and severity of face processing deficits by applying a diverse battery of neuropsychological tests to 45,X"’ and control females. Women with TS performed at below average levels in terms of face and emotion recognition (particularly fearful faces) despite processing faces in a typical configurai manner. Study Two found equivalent deficits in 45,X*^ women. Using Voxel Based Morphometry, Study Three found evidence for increased volume of the amygdalae and orbito-frontal cortices in women with TS. Because males, like 45,X females, have a single X-chromosome, Study Four sought to identify whether there was any sexual dimorphism in face processing abilities - there was not. -

The X's and O's of Turner's Syndrome
Susan Charney Andrea Smillie Copyright ©1987 by Susan Charney Printed in Toronto, Canada First Edition: 1983 Second Edition: 1987 Turner's Syndrome Society of Canada 7777 Keele St. 2nd Floor Concord, Ontario L4K 1Y7 Ph: (905) 660-7766 Fax: (905) 660-7450 Toll Free 1 800 465-6744 This printing has been made possible by a generous donation from Genentech Canada. Acknowledgements The Turner's Syndrome Society would like to thank effort and expertise to the creation and revision o{ this the following Individuals who have contributed their time, booklet: Diane Plumridge for the inspiration and help provided by her booklet Good Things Come In Small Packages. Dr. Johannes Nielsen for his contribution to the sections on physical characteristics and disclosure. Medical Resources J.M. Berg, M.D. C. Cowell, M.D. H.A. Gardner, M.D. E. Greenglass, Ph.D. J. Holland, M.D. I.E. Pearce, Ph.D. J. Rovet, Ph.D. Artistic Contributions Christopher Costello-Cover Karen Fenwick-Drawing Rhonda Reich-Diagrams Editors Michael Bockner Hedy Weiss Special thanks to all those who participated in group discussions, contributed their experiences and sent in their surveys. These individuals helped to make this booklet truly representative of the concerns of those with Turner's Syndrome Introduction The Turner's Syndrome Society of Canada was formed to provide much needed services for those with Turner's Syndrome and their families. The Society held its inaugural meeting in 1981 with only 3 mem bers present. It has since grown to include hundreds of members from all over the world. People with disorders that are not common or well known often feel isolated. -

Technical Details Congential Anomaly Statistics 2017- Technical Details
National Congential Anomaly and Rare Disease Registration Service Congenital anomaly statistics 2017- technical details Congential anomaly statistics 2017- technical details About Public Health England Public Health England exists to protect and improve the nation’s health and wellbeing, and reduce health inequalities. We do this through world-leading science, knowledge and intelligence, advocacy, partnerships and the delivery of specialist public health services. We are an executive agency of the Department of Health and Social Care, and a distinct delivery organisation with operational autonomy. We provide government, local government, the NHS, Parliament, industry and the public with evidence-based professional, scientific and delivery expertise and support. Public Health England Wellington House 133-155 Waterloo Road London SE1 8UG Tel: 020 7654 8000 www.gov.uk/phe Twitter: @PHE_uk Facebook: www.facebook.com/PublicHealthEngland © Crown copyright 2019 You may re-use this information (excluding logos) free of charge in any format or medium, under the terms of the Open Government Licence v3.0. To view this licence, visit OGL. Where we have identified any third party copyright information you will need to obtain permission from the copyright holders concerned. Published August 2019 PHE publications PHE supports the UN gateway number: GW-473 Sustainable Development Goals 2 Congential anomaly statistics 2017- technical details Contents About Public Health England 2 Incidence and birth prevalence 4 Confidence intervals 4 Calculation of birth -

2003 Birmingham
11 S1 The Advanced DNA Banking and European Journal of Human Genetics Isolation Technology – IsoCode® IsoCode is optimized lyse Cells IsoCode is available To isolate DNA Standard Card and Stix archive DNA As ID with colour indicator prepare PCR IsoCode can be used high through-put format identification of individuals you want For population screening forensic testing paternity testing • Supplement 11 1 Volume Archiving DNA, cDNA Clones and Vectors The Official Journal of the European Society of Human Genetics Biological samples dried on IsoCode can be archived indefinitely at ambi- ent temperatures. IsoCode is bactericidal, virucidal, and fungicidal. Isolation of DNA without any reagents EUROPEAN HUMAN GENETICS CONFERENCE 2003 IsoCode binds and inactivates proteins and inhibitors – but not the DNA . DNA is released from IsoCode in a simple water elution process that MAY 3 – 6, 2003, BIRMINGHAM, ENGLAND requires neither reagents nor additional costs. PROGRAMME AND ABSTRACTS High quality DNA – easy and cheap DNA eluted from IsoCode is ready for PCR, automated DNA sequencing, STR analysis, mtDNA analysis and other bio-molecular techniques. Use in a lot of fields May 2003 Ideal for drug discovery SNP libraries, epide- miological studies, human identity appli- cations, forensic samples and as back-up copy. EHGC 2003 Booth 940 Volume 11 – Supplement 1 – May 2003 U.S. Pat. #5.939.259, U.S. Pat. 6.168.922 Made under license from Whatman plc. to U.S. Pat. #5.807.527 Schleicher & Schuell BioScience GmbH · Tel. +49-55 61-79 14 63 · Fax +49-55 61-79 15 83 · D-37582 Dassel · Germany · [email protected] Schleicher & Schuell BioScience Inc. -

EUROCAT Syndrome Guide
JRC - Central Registry european surveillance of congenital anomalies EUROCAT Syndrome Guide Definition and Coding of Syndromes Version July 2017 Revised in 2016 by Ingeborg Barisic, approved by the Coding & Classification Committee in 2017: Ester Garne, Diana Wellesley, David Tucker, Jorieke Bergman and Ingeborg Barisic Revised 2008 by Ingeborg Barisic, Helen Dolk and Ester Garne and discussed and approved by the Coding & Classification Committee 2008: Elisa Calzolari, Diana Wellesley, David Tucker, Ingeborg Barisic, Ester Garne The list of syndromes contained in the previous EUROCAT “Guide to the Coding of Eponyms and Syndromes” (Josephine Weatherall, 1979) was revised by Ingeborg Barisic, Helen Dolk, Ester Garne, Claude Stoll and Diana Wellesley at a meeting in London in November 2003. Approved by the members EUROCAT Coding & Classification Committee 2004: Ingeborg Barisic, Elisa Calzolari, Ester Garne, Annukka Ritvanen, Claude Stoll, Diana Wellesley 1 TABLE OF CONTENTS Introduction and Definitions 6 Coding Notes and Explanation of Guide 10 List of conditions to be coded in the syndrome field 13 List of conditions which should not be coded as syndromes 14 Syndromes – monogenic or unknown etiology Aarskog syndrome 18 Acrocephalopolysyndactyly (all types) 19 Alagille syndrome 20 Alport syndrome 21 Angelman syndrome 22 Aniridia-Wilms tumor syndrome, WAGR 23 Apert syndrome 24 Bardet-Biedl syndrome 25 Beckwith-Wiedemann syndrome (EMG syndrome) 26 Blepharophimosis-ptosis syndrome 28 Branchiootorenal syndrome (Melnick-Fraser syndrome) 29 CHARGE -

Physical Examination of Newborn by Dr Behzad Barekatain MD Assistant Professor of Pediatrics, Neonatologist Academic Member of Isfahan University of Medical Sciences
Physical examination of Newborn By Dr behzad barekatain MD Assistant professor of pediatrics, neonatologist Academic member of isfahan university of medical sciences 1 Swelling of the eyelids is common in newborns and resolves during the first few days of life. Subconjunctival hemorrhages are common. They occur during delivery and resolve in 1 to 2 weeks. 2 The normally white sclera is evaluated for changes of color. A bluish coloration, however, is present in premature infants and in other small babies because of their very thin sclera.also presents in baby with osteogenesis imperfecta. There may also be a mucoid discharge affecting the eyes, often called a “sticky eye,” in the first few days of life, which resolves spontaneously; if more prolonged, it is often from a blocked or incompletely canalized nasolacrimal duct. The eyelids can be cleansed with sterile water. This condition must be contrasted with the erythematous, swollen eyelids with purulent eye discharge seen in conjunctivitis. 4 5 ABNORMAL RED REFLEX This is one of the most important abnormalities that requires immediate evaluation. The term leukocoria is used to describe a white pupil seen by the naked eye or during the red reflex test. Leukocoria is not a diagnosis but rather a description of an observation. Because the infant sleeps much of the time and because the pupils are small, a white pupil often is not noticed until the infant becomes more alert and active. False-positive red reflex test results are commonly due to small pupils, shifting gaze, limited patient cooperation, poor illumination from the ophthalmoscope, and examiner inexperience. -
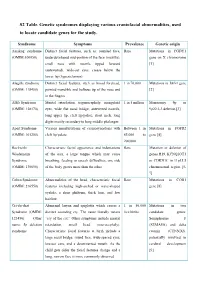
S2 Table. Genetic Syndromes Displaying Various Craniofacial Abnormalities, Used to Locate Candidate Genes for the Study
S2 Table. Genetic syndromes displaying various craniofacial abnormalities, used to locate candidate genes for the study. Syndrome Symptoms Prevalence Genetic origin Aarskog syndrome Distinct facial features, such as: rounded face, Rare Mutations in FGDY1 (OMIM:100050) underdeveloped mid-portion of the face (maxilla), gene on X chromosome small nose with nostrils tipped forward [1] (anteverted), wide-set eyes, crease below the lower lip (hypertelorism) Alagille syndrome Distinct facial features, such as broad forehead, 1 in 70,000 Mutations in JAG1 gene (OMIM: 118450) pointed mandible and bulbous tip of the nose and [2] in the fingers Alfi's Syndrome Mental retardation, trigonocephaly, mongoloid 1 in 5 million Monosomy 9p or (OMIM: 158170) eyes, wide flat nasal bridge, anteverted nostrils, 9p22.2-3 deletion [3] long upper lip, cleft lip/palate, short neck, long digits mostly secondary to long middle phalanges Apert Syndrome Various manifestations of craniosynostosis with Between 1 in Mutations in FGFR2 (OMIM: 101200) cleft lip/palate. 65,000 to gene [4] 200,000 Beckwith- Characteristic facial appearance and indentations Rare Mutation or deletion of Wiedemann of the ears, a large tongue which may cause genes H19, KCNQ1OT1 Syndrome breathing, feeding or speech difficulties, one side or CDKN1C in 11p15.5 (OMIM: 130650) of the body grows more than the other chromosomal region [5- 7] Cohen Syndrome Abnormalities of the head, characteristic facial Rare Mutations in COH1 (OMIM: 216550) features including high-arched or wave-shaped gene [8] eyelids, a short philtrum, thick hair, and low hairline Cri-du-chat Abnormal larynx and epiglottis which causes a 1 in 50,000 Mutations in two Syndrome (OMIM: distinct sounding cry.