Koolen-De Vries Syndrome
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Identification and Characterization of a Novel 43-Bp Deletion Mutation of The
Liu et al. BMC Medical Genetics (2018) 19:61 https://doi.org/10.1186/s12881-018-0567-z CASE REPORT Open Access Identification and characterization of a novel 43-bp deletion mutation of the ATP7B gene in a Chinese patient with Wilson’s disease: a case report Gang Liu†, Dingyuan Ma†, Jian Cheng, Jingjing Zhang, Chunyu Luo, Yun Sun, Ping Hu, Yuguo Wang, Tao Jiang* and Zhengfeng Xu* Abstract Background: Wilson’s disease (WD) is an autosomal recessive disorder characterized by copper accumulation. ATP7B gene mutations lead to ATP7B protein dysfunction, which in turn causes Wilson’s disease. Case presentation: We describe a male case of Wilson’s disease diagnosed at 10 years after routine biochemical test that showed low serum ceruloplasmin levels and Kayser–Fleischer rings in both corneas. Analysis of the ATP7B gene revealed compound heterozygous mutations in the proband, including the reported c.3517G > A mutation and a novel c.532_574del mutation. The c.532_574del mutation covered a 43-bp region in exon 2, and resulted in a frameshift mutation (p.Leu178PhefsX10). By base sequence analysis, two microhomologies (TCTCA) were observed on both deletion breakpoints in the ATP7B gene. Meanwhile, the presence of some sequence motifs associated with DNA breakage near the deletion region promoted DNA strand break. Conclusions: By comparison, a replication-based mechanism named fork stalling and template switching/ microhomology-mediated break-induced replication (FoSTeS/MMBIR) was used to explain the formation of this novel deletion mutation. Keywords: Wilson’s disease, ATP7B, Novel mutation, FoSTeS/MMBIR Background ATP7B protein dysfunction, which in turn causes accumu- Wilson’s disease (WD, OMIM #277900), an autosomal lation of copper in the liver, brain, kidneys and corneas, recessive disorder characterized by abnormal copper ac- with a wide range of clinical symptoms, including hepatic cumulation and related toxicities, is caused by mutations disorders, neuronal degeneration of the brain, and Kayser- in the ATP7B gene (OMIM *606882) [1]. -

Familial Intellectual Disability As a Result of a Derivative Chromosome
Zhang et al. Molecular Cytogenetics (2018) 11:18 DOI 10.1186/s13039-017-0349-x RESEARCH Open Access Familial intellectual disability as a result of a derivative chromosome 22 originating from a balanced translocation (3;22) in a four generation family Kaihui Zhang1†, Yan Huang2†, Rui Dong1†, Yali Yang2, Ying Wang1, Haiyan Zhang1, Yufeng Zhang1, Zhongtao Gai1* and Yi Liu1* Abstract Background: Balanced reciprocal translocation is usually an exchange of two terminal segments from different chromosomes without phenotypic effect on the carrier while leading to increased risk of generating unbalanced gametes. Here we describe a four-generation family in Shandong province of China with at least three patients sharing severe intellectual disability and developmental delay resulting from a derivative chromosome 22 originating from a balanced translocation (3;22) involving chromosomes 3q28q29 and 22q13.3. Methods: The proband and his relatives were detected by using karyotyping, chromosome microarray analysis, fluorescent in situ hybridization and real-time qPCR. Results: The proband, a 17 month-old boy, presented with severe intellectual disability, developmental delay, specific facial features and special posture of hands. Pedigree analysis showed that there were at least three affected patients. The proband and other two living patients manifested similar phenotypes and were identified to have identically abnormal cytogenetic result with an unbalanced translocation of 9.0 Mb duplication at 3q28q29 and a 1.7Mb microdeletion at 22q13.3 by karyotyping and chromosome microarray analysis. His father and other five relatives had a balanced translocation of 3q and 22q. Fluorescence in situ hybridization and real-time qPCR definitely validated the results. -

Nf1 Gene Deletion
NF1 GENE DELETION NF1 GENE DELETION This resource is for families who have a deletion of the NF1 gene causing neurofi bromatosis type 1 (NF1). This is also referred to as NF1 microdeletion. WHAT ARE CHROMOSOMES, DELETION GENES AND MUTATIONS? Chromosomes are the packages of our genetic information. Within each cell of the body are 46 chromosomes arranged in 23 pairs. One chromosome in each pair is inherited from the Source: U.S. National Library of Medicine mother and the other from the father. The pairs are numbered WHAT IS AN NF1 MICRODELETION? by size. The number 1 chromosome When the entire NF1 gene is missing, it is referred pair is the largest and the number 22 is to as NF1 gene deletion or NF1 microdeletion. the smallest. The last pair of chromosomes Approximately 5% of individuals with a diagnosis (sex chromosomes) help to determine whether of NF1 have a deletion that includes the entire NF1 an individual is a male or a female. Genes are gene. Other than the NF1 gene, there are usually small areas along the chromosomes, and are other nearby genes that are also missing. the body’s blueprints or instructions. We have approximately 20,000 genes that control how we WHAT DOES IT MEAN TO HAVE AN NF1 grow and develop and what we look like. Each MICRODELETION? gene can be thought of as a sentence made up of In addition to the NF1 gene, individuals with NF1 four letters (A, T, C and G). Mutations (also called microdeletion typically have other genes in the pathogenic variants), are changes in a gene’s region of chromosome 17 deleted. -

Status of the P53, P16, RB1, and HER-2 Genes and Chromosomes 3
367 ORIGINAL ARTICLE J Clin Pathol: first published as 10.1136/jcp.2004.021154 on 24 March 2005. Downloaded from Status of the p53, p16, RB1, and HER-2 genes and chromosomes 3, 7, 9, and 17 in advanced bladder cancer: correlation with adjacent mucosa and pathological parameters M Gallucci, F Guadagni, R Marzano, C Leonardo, R Merola, S Sentinelli, E M Ruggeri, R Cantiani, I Sperduti, F de la Iglesia Lopez, A M Cianciulli ............................................................................................................................... J Clin Pathol 2005;58:367–371. doi: 10.1136/jcp.2004.021154 Aims: To evaluate a panel of well known genetic alterations for frequency of changes in bladder cancer that could be considered genomic instability determinants or adjunctive prognostic predictors. Methods: Fluorescence in situ hybridisation analysis was performed to evaluate chromosomes 3, 7, 9, and 17 and the 9p21 (p16), 17p13.1 (p53), 13q14 (RB1), and 17q11.2 (HER-2) chromosomal loci in 48 See end of article for muscle invasive bladder cancer specimens and the adjacent normal mucosa. authors’ affiliations Results: There were significant differences between the frequency of chromosome 7 monosomy/polysomy ....................... and 17 monosomy in the two groups (tumours and adjacent mucosa) (p = 0.004, p = 0.037, and Correspondence to: p = 0.015, respectively). There were no differences in the frequency of gene deletions between tumours Dr A M Cianciulli, Clinical and the adjacent mucosa. 17q11.2 amplification was found in 14.5% of tumours examined, but not in the Pathology, Regina Elena non-malignant epithelium. Chromosome 3, 7, and 17 monosomy and the RB1 heterozygous deletion were Cancer Institute, IFO, Via Elio Chianesi, 53, 00144 significantly associated with stage T3–4 (p = 0.03, p = 0.04, p = 0.04, and p = 0.03, respectively). -

Koolen-De Vries Syndrome: Clinical Report of an Adult and Literature Review
Case Report Cytogenet Genome Res 2016;150:40–45 Accepted: July 25, 2016 DOI: 10.1159/000452724 by M. Schmid Published online: November 17, 2016 Koolen-de Vries Syndrome: Clinical Report of an Adult and Literature Review Claudia Ciaccio Chiara Dordoni Marco Ritelli Marina Colombi Division of Biology and Genetics, Department of Molecular and Translational Medicine, School of Medicine, University of Brescia, Brescia , Italy Key Words Koolen-de Vries syndrome (KdS, also known as 17q21.31 · Deletion · Joint hypermobility · KANSL1 17q21.31 microdeletion syndrome, OMIM #610443) is a rare genetic disorder (prevalence 1/16,000) characterized by typical facial dysmorphisms, cardiac and renal defects, Abstract developmental delay, and intellectual disability of vari- Koolen-de Vries syndrome (KdS) is a rare genetic condition able level [Tan et al., 2009]. The disorder was initially de- characterized by typical facial dysmorphisms, cardiac and re- scribed as a form of mental retardation caused by a 440– nal defects, skeletal anomalies, developmental delay, and in- 680-kb deletion in the 17q21.31 region, typically encom- tellectual disability of variable level. It is caused by a 440– passing 5 genes: CRHR1 (OMIM 122561), MAPT 680-kb deletion in the 17q21.31 region, encompassing (OMIM 157140), IMP5 (OMIM 608284), STH (OMIM CRHR1 , MAPT , IMP5 , STH , and KANSL1 , or by an intragenic 607067), and KANSL1 (OMIM 612452)* [Koolen et al., KANSL1 mutation. The majority of the patients reported are 2006]. Recently,* it has been shown* that haploinsufficien- pediatric or young adults, and long-term studies able to de- cy* of KANSL1 by itself, due to single* nucleotide variants fine the prognosis of the disease are lacking. -

3 Chromosome Chapter
Chromosome 3 ©Chromosome Disorder Outreach Inc. (CDO) Technical genetic content provided by Dr. Iosif Lurie, M.D. Ph.D Medical Geneticist and CDO Medical Consultant/Advisor. Ideogram courtesy of the University of Washington Department of Pathology: ©1994 David Adler.hum_03.gif Introduction The size of chromosome 3 is ~200 Mb. Within this chromosome, there are thousands of genes, many of which are necessary for normal intellectual development or involved in the formation of body organs. Deletions of Chromosome 3 The length of the short arm of chromosome 3 is ~90 Mb. Most known deletions of 3p are caused by the loss of its distal 15 Mb segment (3p25–pter). Deletions of the more proximal segments are relatively rare; there are only ~50 reports on such patients. Therefore, it would be premature to talk about any syndrome related to deletions of the proximal part of 3p. The location of the breakpoints, size of deletion, and reported abnormalities are different in most described patients. However, recurrent aortal stenosis in patients with deletion 3p11p14.2, abnormal lung lobation in patients with deletion 3p12p14.2, agenesis or hypoplasia of the corpus callosum in patients with deletion 3p13, microphthalmia and coloboma in patients with deletion 3p13p21.1, choanal atresia and absent gallbladder in patients with deletion 3p13p21, and hearing loss in patients with deletion 3p14 are all indicators that the above–mentioned segments likely contain genes involved in the formation of these systems. Deletions of 3p Deletion of 3p25–pter The most distal segment of the short arm of chromosome 3 is 3p26 and spans ~8 Mb. -

Trisomy 5P Inverted Duplication & Deletion of 5Pftnwdraft3
Trisomy 5p: Inverted duplication and deletion of 5p rarechromo.org Inverted duplication with deletion of 5p Inverted duplication with deletion of 5p, known as inv dup del 5p, is a very rare genetic condition in which there is an extra copy of part of the genetic material (DNA) that makes up the body’s 46 chromosomes, and a missing copy of another part. Like most other chromosome disorders, this usually affects development, and sometimes health and behaviour as well. It is likely that both the extra and missing parts of chromosome 5p have an effect, but a lot depends on their position and size. The precise effects of gaining material from a chromosome vary depending on how large the duplication is, how many genes it contains and what those genes do. The same applies to deletions. The effects may not be limited to the genes within the duplicated or deleted piece of chromosome because these genes may interact with other genes on the same chromosome or other chromosomes. Chromosomes usually come in pairs, and we inherit one chromosome from each parent. Of the 46 chromosomes, two are a pair of sex chromosomes: two Xs for a girl and an X and a Y for a boy. The remaining 44 chromosomes are grouped into 22 pairs and are numbered 1 to 22, approximately from largest to smallest. Each chromosome has a short (p) arm (from petit, the French for small) and a long (q) arm. The diagram below shows the short arm. Chromosome 5 Short (p) arm Bands Base pairs 0Mb 5Mb 10Mb 15Mb 20Mb 25Mb 30Mb 35Mb 40Mb 45Mb 48.4Mb Long (q) arm 2 People have 2 copies of chromosome 5 in most of their body cells. -

Early Fetal Presentation of Koolen-De Vries: Case Report with Literature Review
European Journal of Medical Genetics xxx (2017) 1e5 Contents lists available at ScienceDirect European Journal of Medical Genetics journal homepage: http://www.elsevier.com/locate/ejmg Early fetal presentation of Koolen-de Vries: Case report with literature review abstract Keywords: Koolen-de Vries syndrome (MIM#610443) is a rare microdeletion syndrome involving the 17q21.31 Koolen-de Vries syndrome region, which was first described by Koolen in 2006. Clinical and behavioral characteristics have been 17q21.31 deletion extensively reported from more than 100 postnatal cases including infants, children and young adults. Prenatal array-CGH The syndrome is highly clinically heterogeneous, but the main features associate characteristic cranio- Neuropathology facial dysmorphism, heart defects, limb, skeletal, genito-urinary anomalies, along with intellectual Cytogenetics Mega corpus callosum disability with early childhood epilepsy and behavioral disturbances. Central nervous system malfor- mations usually consist in hydrocephalus and thin corpus callosum. We report herein an early fetal case with an apparently isolated abnormal corpus callosum diagnosed by ultrasonography, for which a medical termination of the pregnancy was achieved at 22 weeks of gestation. Postmortem examination displayed facial dysmorphism consisting of hypertelorism, short philtrum and flat and broad nose, cleft palate and left duplex ureter. Neuropathological examination revealed a mega corpus callosum that has never been reported so far in this syndrome. Array-CGH performed -

The Deletion Stocks of Common Wheat
The Deletion Stocks of Common Wheat T. R. Endo and B. S. Gill Chromosomal breaks occurred in the progeny of a common wheat (Tritlcum aes- tlvum L. em Thell; 2n = 6x = 42, genome formula AABBDD) cultivar Chinese Spring with a monosomic addition of an alien chromosome from Aegllops cyllndrlca Host (2n = 4x = 28, CCDD) or A. trlunclalls L. (2n = 4x = 28, UUCC) or a chromosomal segment from A. spettoldes Tausch (2n = 2x = 14, SS). We identified 436 deletions by C-banding. The deletion chromosomes were transmitted stably to the offspring. We selected deletion homozygotes in the progeny of the deletion heterozygotes and established homozygous lines for about 80% of the deletions. We failed to establish homozygous lines for most of the deletions In the short arm of chromo- some 2A and for all deletions in the short arm of chromosome 4B, because plants homozygous for these deletions were sterile. We could not obtain any homozygotes for larger deletions In the long arms of chromosomes 4A, 5A, 5B, and 5D. The deletion stocks showed variations in morphological, physiological, and biochemi- cal traits, depending on the size of their chromosomal deficiency, and are powerful tools for physical mapping of wheat chromosomes. The aneuploid stocks developed in a com- and a chromosome fragment from A. spel- mon wheat cultivar Chinese Spring are a toides were found recently to induce chro- powerful tool for genetic and breeding mosome mutations in Chinese Spring in studies of wheat (Sears 1954, 1966; Sears the same manner as the A. cylindrica chro- and Sears 1978). These stocks are im- mosome (Endo, unpublished data). -

A Rare and Unusual Case of Trisomy 10P with Terminal 14Q Deletion: a Multidisciplinary Approach
Open Access Case Report DOI: 10.7759/cureus.15459 A Rare and Unusual Case of Trisomy 10p with Terminal 14q Deletion: A Multidisciplinary Approach Chanan Goyal 1, 2 , Vivek Goyal 3 , Waqar M. Naqvi 1 1. Community Physiotherapy, Datta Meghe Institute of Medical Sciences, Wardha, IND 2. Paediatric Physiotherapy, Government Physiotherapy College, Raipur, IND 3. Anaesthesiology, Shri Balaji Institute of Medical Science, Raipur, IND Corresponding author: Chanan Goyal, [email protected] Abstract Trisomy 10p is a rare entity to be diagnosed and so is terminal 14q deletion. The total number of trisomy 10p cases reported to date is estimated to be in double digits. The number of terminal 14q deletion cases that have been reported in the literature is even lesser than that of trisomy 10p. Simultaneous occurrence of these genetic aberrations is, therefore, extremely rare. Herein, we document a case of a 14-month-old female diagnosed with trisomy 10p and terminal 14q deletion, who presented with an inability to sit without support and had difficulty in holding her neck. She had no means of independent indoor mobility, which was further limiting her development by exploration. Clinical features included hypotonia, developmental delay, extraneous movements of the head and tongue, intellectual impairment, and facial dysmorphism. She could maintain tripod sitting for less than a minute. Physiotherapy intervention was based on principles of neurodevelopmental treatment and sensory integration. After nine months of physiotherapy intervention, her total gross motor function measure (GMFM) score improved from 11% to 40%. The functional gains were maintained with a home exercise program, after almost one year of discontinuation of institution-based physiotherapy. -

Microduplications of 22Q11.2 Are Frequently Inherited and Are Associated with Variable Phenotypes Zhishuo Ou, MD1, Jonathan S
April 2008 ⅐ Vol. 10 ⅐ No. 4 article Microduplications of 22q11.2 are frequently inherited and are associated with variable phenotypes Zhishuo Ou, MD1, Jonathan S. Berg, MD, PhD1, Hagith Yonath, MD1, Victoria B. Enciso, MS2, David T. Miller, MD, PhD3, Jonathan Picker, MD3, Tiffanee Lenzi, MD, PhD4, Catherine E. Keegan, MD, PhD4, Vernon R. Sutton, MD1,5, John Belmont, MD, PhD1,5, A. Craig Chinault, PhD1, James R. Lupski, MD, PhD1,5,6, Sau Wai Cheung, PhD, MBA1, Elizabeth Roeder, MD2, and Ankita Patel, PhD1 Purpose: Genomic rearrangements of chromosome 22q11.2, including the microdeletion associated with Di- George/velocardiofacial syndrome, are mediated by nonallelic homologous recombination between region-specific low-copy repeats. To date, only a small number of patients with 22q11.2 microduplication have been identified. Methods: We report the identification by array-comparative genomic hybridization of 14 individuals from eight families who harbor microduplications within the 22q11.2 region. Results: We have now observed a variety of microduplications, including the typical common ϳ3-Mb microduplication, ϳ1.5-Mb nested duplication, and smaller microduplications within and distal to the DiGeorge/velocardiofacial syndrome region, consistent with nonallelic homologous recombination using distinct low-copy repeats in the 22q11.2 DiGeorge/velocardiofacial syndrome region. These microduplications likely represent the predicted reciprocal rearrangements to the microde- letions characterized in the 22q11.2 region. The phenotypes seen in these individuals are generally mild and highly variable; familial transmission is frequently observed. Conclusions: These findings highlight the unbiased ability of array-comparative genomic hybridization to identify genomic imbalances and further define the molecular etiology and clinical phenotypes seen in microduplication 22q11.2 syndrome. -
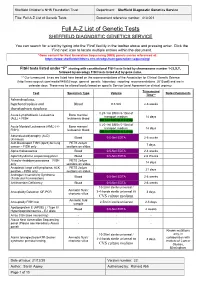
Full A-Z List of Genetic Tests Document Reference Number: 413.001
Sheffield Children’s NHS Foundation Trust Department: Sheffield Diagnostic Genetics Service Title: Full A-Z List of Genetic Tests Document reference number: 413.001 Full A-Z List of Genetic Tests SHEFFIELD DIAGNOSTIC GENETICS SERVICE You can search for a test by typing into the ‘Find’ facility in the toolbar above and pressing enter. Click the ‘Find next’ icon to locate multiple entries within the document. *Gene content for Next Generation Sequencing (NGS) panels can be referenced at: https://www.sheffieldchildrens.nhs.uk/sdgs/next-generation-sequencing/ FISH tests listed under “F” starting with constitutional FISH tests listed by chromosome number 1-22,X,Y, followed by oncology FISH tests listed A-Z by gene name. ** Our turnaround times are listed here based on the recommendations of the Association for Clinical Genetic Science (http://www.acgs.uk.com/media/949852/acgs_general_genetic_laboratory_reporting_recommendations_2015.pdf) and are in calendar days. These may be altered locally based on specific Service Level Agreement or clinical urgency. Turnaround Test Specimen Type Volume Notes/Comments Time** Achondroplasia, hypchondroplasia and Blood 0.5-5ml 2-6 weeks thanatophoric dysplasia 0.25-1ml BM in 5-10ml of Acute Lymphoblastic Leukaemia Bone marrow/ transport medium 14 days (ALL) + FISH leukaemic blood OR 1ml BM/VB in Li Hep 0.25-1ml BM in 5-10ml of Acute Myeloid Leukaemia (AML) (+/- Bone marrow/ transport medium 14 days FISH) leukaemic blood OR 1ml BM/VB in Li Hep Adrenoleukodystrophy (ALD) Blood 0.5-5ml EDTA 2-6 weeks (X-linked)