G-33-605 90999.Pdf
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-
Densifying Metal Hydrides with High Temperature and Pressure
3,784,682 United States Patent Office Patented Jan. 8, 1974 feet the true density. That is, by this method only theo- 3,784,682 retical or near theoretical densities can be obtained by DENSIFYING METAL HYDRIDES WITH HIGH making the material quite free from porosity (p. 354). TEMPERATURE AND PRESSURE The true density remains the same. Leonard M. NiebylsM, Birmingham, Mich., assignor to Ethyl Corporation, Richmond, Va. SUMMARY OF THE INVENTION No Drawing. Continuation-in-part of abandoned applica- tion Ser. No. 392,370, Aug. 24, 1964. This application The process of this invention provides a practical Apr. 9,1968, Ser. No. 721,135 method of increasing the true density of hydrides of Int. CI. COlb 6/00, 6/06 metals of Groups II-A, II-B, III-A and III-B of the U.S. CI. 423—645 8 Claims Periodic Table. More specifically, true densities of said 10 metal hydrides may be substantially increased by subject- ing a hydride to superatmospheric pressures at or above ABSTRACT OF THE DISCLOSURE fusion temperatures. When beryllium hydride is subjected A method of increasing the density of a hydride of a to this process, a material having a density of at least metal of Groups II-A, II-B, III-A and III-B of the 0.69 g./cc. is obtained. It may or may not be crystalline. Periodic Table which comprises subjecting a hydride to 15 a pressure of from about 50,000 p.s.i. to about 900,000 DESCRIPTION OF THE PREFERRED p.s.i. at or above the fusion temperature of the hydride; EMBODIMENT i.e., between about 65° C. -

New Generation Cadmium‑Free Quantum Dots for Biophotonics and Nanomedicine
This document is downloaded from DR‑NTU (https://dr.ntu.edu.sg) Nanyang Technological University, Singapore. New Generation Cadmium‑Free Quantum Dots for Biophotonics and Nanomedicine Xu, Gaixia; Zeng, Shuwen; Zhang, Butian; Swihart, Mark T.; Yong, Ken‑Tye; Prasad, Paras N. 2016 Xu, G., Zeng, S., Zhang, B., Swihart, M. T., Yong, K.‑T., & Prasad, P. N. (2016). New Generation Cadmium‑Free Quantum Dots for Biophotonics and Nanomedicine. Chemical Reviews, 116(19), 12234‑12327. https://hdl.handle.net/10356/83973 https://doi.org/10.1021/acs.chemrev.6b00290 © 2016 American Chemical Society. This is the author created version of a work that has been peer reviewed and accepted for publication by Chemical Reviews, American Chemical Society. It incorporates referee’s comments but changes resulting from the publishing process, such as copyediting, structural formatting, may not be reflected in this document. The published version is available at: [http://dx.doi.org/10.1021/acs.chemrev.6b00290]. Downloaded on 06 Oct 2021 00:28:06 SGT Page 1 of 288 Submitted to Chemical Reviews 1 2 3 4 5 6 New Generation Cadmium-Free Quantum Dots for 7 8 Biophotonics and Nanomedicine 9 10 11 12 13 14 Gaixia Xu1,5†, Shuwen Zeng2,5†, Butian Zhang2, Mark T. Swihart3*, 15 2* 4* 16 Ken-Tye Yong and Paras N. Prasad 17 18 19 20 1 21 Key Laboratory of Optoelectronics Devices and Systems of Ministry of Education/Guangdong 22 Province, College of Optoelectronic Engineering, Shenzhen University, Shenzhen, P. R. 23 China 24 2 25 School of Electrical and Electronic Engineering, -

S-Block Amidoboranes: Syntheses, Structures, Reactivity and Applications Cite This: Chem
Chem Soc Rev View Article Online REVIEW ARTICLE View Journal | View Issue s-Block amidoboranes: syntheses, structures, reactivity and applications Cite this: Chem. Soc. Rev., 2016, 45, 1112 Tom E. Stennett and Sjoerd Harder* Metal amidoborane compounds of the alkali- and alkaline earth metals have in recent years found applications in diverse disciplines, notably as hydrogen storage materials, as reagents for the reduction of organic functional groups and as catalysts and intermediates in dehydrocoupling reactions. These functions are connected by the organometallic chemistry of the MNR2BH3 group.† This review focusses on central aspects of the s-block amidoborane compounds – their syntheses, structures and reactivity. Well-defined amidoborane complexes of group 2 metals are now available by a variety of solution-phase routes, which has allowed a more detailed analysis of this functional group, which was previously largely confined to solid-state materials chemistry. Structures obtained from X-ray crystallography have begun to provide increased understanding of the fundamental steps of key processes, including amine–borane Creative Commons Attribution 3.0 Unported Licence. dehydrocoupling and hydrogen release from primary and secondary amidoboranes. We review structural parameters and reactivity to rationalise the effects of the metal, nitrogen substituents and supporting Received 10th July 2015 ligands on catalytic performance and dehydrogenative decomposition routes. Mechanistic features of DOI: 10.1039/c5cs00544b key processes involving amidoborane compounds as starting materials or intermediates are discussed, alongside emerging applications such as the use of group 1 metal amidoboranes in synthesis. Finally, the www.rsc.org/chemsocrev future prospects of this vibrant branch of main group chemistry are evaluated. -

Daisy Brook Dispatch
Daisy Brook Elementary Daisy Brook Dispatch Volume 1, Issue 2 OCTOBER/NOVEMBER 2007 Inside this issue: A Time to Reflect MEAP Testing Finished 2 Dear Daisy Brook Friends, projects Daisy Brook Fall Book Fair 2 Portable Computer Lab 2 As we start this busy holiday sea- • A school board and adminis- son I hope that we can take the trative staff who are in it “for Portable Computer Lab Con- 3 time to reflect on the many things kids” and not for the politics Mrs. Heft’s Class 4 that we have to be thankful for. of education Halloween Party The list for each of us is different but I would like to take a minute As you continue to read through this Mrs. Hanna’s Class Trip 4 and list some of the positive influ- newsletter take note of the many Daisy Brook Marathon 5 ences that affect our educational opportunities that our students learning environment at Daisy have. It is not the same in all Daisy Brook Cross 6-9 Brook. districts. We are truly blessed. Country Run Counselor’s Corner 9 • Supportive parent group that The Daisy Brook Staff and I wish Before/After School 9 just completed a wonderful you the best of this holiday season. Traffic fund raiser for our students Sincerely, Indoor/Outdoor Recess 10 Guidelines • A great staff of teachers who are dedicated to the love of Weather Guidelines for 10 learning School Closing/Delays • A community that cares What’s Happening 11 about its kids and wants Nancy Sparks, Principal them to be successful Daisy Brook School Color Me 12 Pine Street Primary Center • Mrs. -

Hazardous Laboratory Chemicals Disposal Guide
Third Edition HAZARDOUS LABORATORY CHEMICALS DISPOSAL GUIDE Third Edition HAZARDOUS LABORATORY CHEMICALS DISPOSAL GUIDE Margaret-Ann Armour LEWIS PUBLISHERS A CRC Press Company Boca Raton London New York Washington, D.C. This edition published in the Taylor & Francis e-Library, 2005. To purchase your own copy of this or any of Taylor & Francis or Routledge’s collection of thousands of eBooks please go to http://www.ebookstore.tandf.co.uk/. Library of Congress Cataloging-in-Publication Data Armour, M.A. (Margaret-Ann) Hazardous laboratory chemicals disposal guide/Margaret-Ann Armour.—3rd ed. p. cm. Includes bibliographical references. ISBN 1-56670-567-3 1. Chemical laboratories—Waste disposal. 2. Hazardous substances. I. Title. QD64.A76 2003 542′.89–dc21 2002043358 This book contains information obtained from authentic and highly regarded sources. Reprinted material is quoted with permission, and sources are indicated. A wide variety of references are listed. Reasonable efforts have been made to publish reliable data and information, but the authors and the publisher cannot assume responsibility for the validity of all materials or for the consequences of their use. Neither this book nor any part may be reproduced or transmitted in any form or by any means, electronic or mechanical, including photocopying, microfilming, and recording, or by any information storage or retrieval system, without prior permission in writing from the publisher. The consent of CRC Press LLC does not extend to copying for general distribution, for promotion, for creating new works, or for resale. Specific permission must be obtained in writing from CRC Press LLC for such copying. Direct all inquiries to CRC Press LLC, 2000 N.W. -

Flexible Solar Cells
Flexible Solar Cells A Major Qualifying Project Submitted to the Faculty Of Worcester Polytechnic Institute In Partial Fulfillment of the requirements for the Degree in Bachelor of Science In Mechanical Engineering By Francis LaRovere Edward Peglow Michael McMahon Project Advisor Professor Pratap Rao, Advisor This report represents work of WPI undergraduate students submitted to the faculty as evidence of a degree requirement. WPI routinely publishes these reports on its w ebsite without editorial or peer review. For more information about the projects program at WPI, see http://www.wpi.edu/Academics/Projects. 1 Table of Contents Table of Figures ............................................................................................................................................. 5 Table of Tables .............................................................................................................................................. 6 Acknowledgments ......................................................................................................................................... 7 Abstract ......................................................................................................................................................... 8 1. Introduction .............................................................................................................................................. 9 2. Scope ...................................................................................................................................................... -

UNITED STATES PATENT OFFICE 2,56,31 METHOD of REDUCING and by DRO GENATING CHEMICA, COMPOUNDS by REACTING WITE: ALUMNUM-CONAN NG BYOFREDES Hermann E
Patented Nov. 27, 1951 2,576,31 UNITED STATES PATENT OFFICE 2,56,31 METHOD OF REDUCING AND BY DRO GENATING CHEMICA, COMPOUNDS BY REACTING WITE: ALUMNUM-CONAN NG BYOFREDES Hermann E. Schlesirager and Albert E. Finholt, Chicago, Ill.; said Schlesinger assignor of one fourth to. Edaa, M. Schlesinger and said Fin holt assignor of one-fourth to Marion H. Finholt No Drawing. Application June 3, 1947, Serial No. 752,286 2 (Cairns. (C. 260-638) 2 This invention relates to methods of making LiAlH4. Although this new compound will be aluminum-containing hydrides and the reactions called lithium aluminum hydride in the present thereof, and also relates to products prepared by application, it may also be called lithium alumi said methods. nohydride or lithium tetrahydroaluminide. In This application is a continuation-in-part of one method of making lithium aluminum hydride, our copending application Serial No. 717,312, filed lithium hydride is reacted with an aluminum December 19, 1946, now Patent No. 2,567,972, halide such as aluminum chloride in the presence issued September 18, 1951. of a suitable liquid medium such as an ether. If We have discovered that these compounds, es the reagents are mixed in the proportions of the pecially the ether soluble lithium aluminum hy 0 following equation, or if an excess of lithium hy dride, are extremely useful chemical reagents. dride is used, the reaction proceeds as follows: - They may be employed for replacing halogens or Organic radicals by hydrogen in a great variety 4Li H--AlCl3->LiAlH4--3LiCl of compounds. As a result, their discovery has led to new methods, safer, more convenient, and 16 The liquid medium used is one in which one of more efficient than those hitherto known, for pro the reaction products, e. -
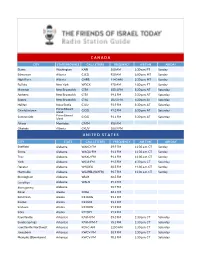
C a N a D a U N I T E D S T a T
C A N A D A CITY STATE/PROVINCE CALL LETTERS FREQUENCY AIR TIME AIR DAY Blaine Washington KARI 550 AM 1:30 a.m. PT Sunday Edmonton Alberta CJCD 930 AM 6:00 p.m. MT Sunday High River Alberta CHRB 1140 AM 2:30 p.m. MT Sunday Buffalo New York WDCX 970 AM 1:00 p.m. ET Sunday Moncton New Brunswick CITA 105.1 FM 5:30 p.m. AT Saturday Amherst New Brunswick CITA 99.1 FM 5:30 p.m. AT Saturday Sussex New Brunswick CITA 107.3 FM 5:30 p.m. AT Saturday Halifax Nova Scotia CJLU 93.9 FM 5:30 p.m. AT Saturday Charlottetown Prince Edward CIOG 91.3 FM 5:30 p.m. AT Saturday Island Summerside Prince Edward CIOG 91.1 FM 5:30 p.m. AT Saturday Island Altona Manitoba CFAM 950 AM Okotoks Alberta CKUV 100.9 FM U N I T E D S T A T E S CITY STATE CALL LETTERS FREQUENCY AIR TIME AIR DAY Sheffield Alabama WAKD-FM 89.9 FM 11:30 a.m. CT Sunday Selma Alabama WAQU-FM 91.1 FM 11:30 a.m. CT Sunday Troy Alabama WAXU-FM 91.1 FM 11:30 a.m. CT Sunday York Alabama WSJA-FM 91.3 FM 4:30 p.m. CT Saturday Decatur Alabama W203DJ 88.5 FM 11:30 a.m. CT Sunday Huntsville Alabama W229BL (WAFR) 93.7 FM 11:30 a.m. CT Sunday Birmingham Alabama WLJR 88.5 FM Carrollton Alabama WALN 89.3 FM Montgomery Alabama 92.7 FM Kenai Alaska KOGJ 88.1 FM Ketchikan Alaska K216DG 91.1 FM Kodiak Alaska K216DF 91.1 FM Seldovia Alaska K220FW 91.9 FM Sitka Alaska K220FY 91.9 FM Fayetteville Arkansas KAYH-FM 89.3 FM 1:30 p.m. -

Seattle a Digital Community Still in Transition Jessica Durkin, Tom Glaisyer, and Kara Hadge, Media Policy Initiative June 2010, Release 2.0
New America Foundation An Information Community Case Study: Seattle A digital community still in transition Jessica Durkin, Tom Glaisyer, and Kara Hadge, Media Policy Initiative June 2010, Release 2.0 Seattle, Washington, could be considered a city singularly suited to develop a healthy democracy in the digital age. The city government, citizens and business have created a productive environment for the next generation of information-sharing and community engagement. Years of economic growth and relative prosperity have fostered new, superior practices in news and information. Yet, losing a major print newspaper, as Seattle did when The Seattle Post-Intelligencer closed, adversely affects a community, by leaving it with one less place to provide public service journalism, stories about people and general community updates. In parallel, Seattle has been at the center of an explosion of alternative news outlets, especially online, which has created a critical mass of information portals for geographic and social communities. As the Knight Report, Informing Communities: Sustaining Democracy in a Digital Age, highlights, it is important to understand that there are three important elements to be considered as we analyze media and democracy in the 21st century: • availability of relevant and credible information to all Americans and their communities; • capacity of individuals to engage with information; and • individual engagement with information and the public life of the community. However, despite the relative vibrancy of the media scene, and even with all its demographic and other advantages, it is unclear how much of this innovation is sustainable. The local web is littered with websites that are no longer updated, and few of the startups boast anything like the journalistic firepower or profitability of the papers of the past. -

UNIVERSITY of CALIFORNIA, IRVINE Transition Metal Complexes
UNIVERSITY OF CALIFORNIA, IRVINE Transition Metal Complexes of Modified Azaphosphatranes DISSERTATION submitted in partial satisfaction of the requirements for the degree of DOCTOR OF PHILOSOPHY in Chemistry by Zachary Thammavongsy Dissertation Committee: Professor Jenny Y. Yang, Chair Professor Andrew S. Borovik Professor William J. Evans 2019 © 2019 Zachary Thammavongsy DEDICATION For my mother, Kor, and my father, Vinai, who sacrificed their lives for me to live out mine. ii TABLE OF CONTENTS Page LIST OF FIGURES vii LIST OF TABLES xv LIST OF SCHEMES xvi LIST OF CHARTS xvii ACKNOWLEDGMENTS xviii CURRICULUM VITAE xix ABSTRACT OF THE DISSERTATION xxv INTRODUCTION 1 CHAPTER 1: The Electronic and Steric Tolman Parameters of Proazaphosphatranes: Synthesis, Characterization, and Measurements 12 1.1. Motivation and Specific Aims 13 1.2. Background 13 1.3. Results and Discussion 15 R 1.3.1. Synthesis and Structure of Ni(L )(CO)3 Complexes (1-4) 15 Me 1.3.2. Synthesis and Structure of Ni(L )2(CO)2 Complex (5) 18 R 1.3.3. Tolman Electronic Parameters and Cone Angles of Ni(L )(CO)3 Complexes (1-4) 21 1.4. Conclusion 23 1.5. Experimental Details 24 1.6. References 38 CHAPTER 2: Expanding the Denticity of Proazaphosphatrane: Ligand Synthesis 41 2.1. Motivation and Specific Aims 42 2.2. Background 43 2.3. Results and Discussion 43 2.3.1. Synthesis of Tri-Substituted Tris(2-aminoethyl)amines 43 2.3.2. Synthesis of Protonated Tri-Substituted Azaphosphatranes 44 2.3.3. Synthesis of Tri-Substituted Proazaphosphatranes 46 2.4. Conclusion 49 2.5. -

Preparations, Solution Composition, and Reactions of Complex Metal Hydrides and Ate Complexes of Zinc, Aluminum, and Copper a Th
PREPARATIONS, SOLUTION COMPOSITION, AND REACTIONS OF COMPLEX METAL HYDRIDES AND ATE COMPLEXES OF ZINC, ALUMINUM, AND COPPER A THESIS Presented to The Faculty of the Division of Graduate Studies By John Joseph Watkins In Partial Fulfillment of the Requirements for the Degree Doctor of Philosophy in the School of Chemistry Georgia Institute of Technology April, 1977 PREPARATIONS, SOLUTION COMPOSITION, AND REACTIONS OF COMPLEX METAL HYDRIDES AND ATE COMPLEXES OF ZINC, ALUMINUM, AND COPPER Approved: Erlin^rbrovenstein, Jr., Chairman H. 0. House E. C. Ashby Date approved by Chairman £~^3c>l~l~f ii ACKNOWLEDGMENTS Many individuals and organizations have contributed to the successful completion of this thesis. The following acknowledgments are not complete, but I hope I have expressed my gratitude to the people and organizations upon whom I depended the most. The School of Chemistry supported my first three years of work by the award of an NSF fellowship. My last year of work was generously supported by the St. Regis Paper Company, who graciously gave me leave of absence with salary so that the requirements for this thesis could be completed. This stipend and tuition support of my work freed me to concentrate on research without the financial difficulties encountered by many graduate students. All the faculty and staff of the School of Chemistry supported my research. I particularly would like to recognize Professor W. M. Spicer, Professor J. A. Bertrand, Professor C. L. Liotta, Mr. Gerald O'Brien, and Mr. D. E. Lillie. Post-doctoral assistants and fellow graduate students who contributed to my experience at the Georgia Insti tute of Technology include Dr. -

Reversible Coordination of Boron−, Aluminum−, Zinc−, Magnesium
Forum Article pubs.acs.org/IC Reversible Coordination of Boron−, Aluminum−, Zinc−, − − Magnesium , and Calcium Hydrogen Bonds to Bent {CuL2} Fragments: Heavy σ Complexes of the Lightest Coinage Metal Alexandra Hicken,†,‡ Andrew J. P. White,‡ and Mark R. Crimmin*,‡ † ‡ SSCP DTP, Grantham Institute, and Department of Chemistry, Imperial College London, South Kensington, London SW7 2AZ, U.K. *S Supporting Information ABSTRACT: A series of copper(I) complexes bearing electron-deficient β- diketiminate ligands have been prepared. The study includes [{{ArNC(CR3)}2CH}- η2 · · Cu( -toluene)n] (Ar = Mes, R = F, n = 0.5, [12 tol]; Ar = C6F5, R = Me, n =1,[2 · − · tol]; Ar = 2,6-Cl2C6H3,R=H,n = 0.5, [32 tol]). Reactions of [1 3n tol] with boranes, alanes, a zinc hydride, a magnesium hydride, and a calcium hydride generate the corresponding σ complexes ([1−3·B], [3·B′], [3·Al], [3·Al′], [1−3·Zn], [1·Mg], and [1·Ca]). These species all form reversibly, being in equilibrium with the arene solvates in solution. With the exception of the calcium complex, the complexes have all been characterized by single-crystal X-ray diffraction studies. In solution, the σ-hydride of the aluminum, zinc, magnesium, and − − − σ calcium derivatives resonates between 0.12 and 1.77 ppm (C6D6 or toluene-d8, 193 298 K). For the -borane complexes, the hydrides are observed as a single resonance between 2 and 3.5 ppm (C6D6, 298 K) and bridging and terminal hydrides rapidly exchange on the NMR time scale even at 193 K. Quantification of the solution dynamics by van’tHoff analysis yields expectedly small values of ΔH° and negative values of ΔS° consistent with weak binding and a reversible process that does not involve aggregation of the copper species.