Genetic Tests of Biologic Systems in Affective Disorders
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Recombinant Human Phosphodiesterase 4A/PDE4A
Recombinant Human Phosphodiesterase 4A/PDE4A Catalog Number: 7767-PE DESCRIPTION Source Spodoptera frugiperda, Sf 21 (baculovirus)derived Pro331Met723, with an Nterminal Met and a Cterminal 6His tag Accession # P27815 Nterminal Sequence Pro331 Analysis Predicted Molecular 46 kDa Mass SPECIFICATIONS SDSPAGE 4448 kDa, reducing conditions Activity Measured by its ability to convert cAMP to 5'AMP. The specific activity is >28,000 pmol/min/μg, as measured under the described conditions. Endotoxin Level <0.01 EU per 1 μg of the protein by the LAL method. Purity >95%, by SDSPAGE under reducing conditions and visualized by Colloidal Coomassie® Blue stain at 5 μg per lane. Formulation Supplied as a 0.2 μm filtered solution in Tris and NaCl. See Certificate of Analysis for details. Activity Assay Protocol Materials l Assay Buffer (1X): 20 mM Tris, 1 mM MgCl2, 1 mM DTT, 0.01538% CHAPS, pH 7.5 l Recombinant Human Phosphodiesterase 4A/PDE4A (rhPDE4A) (Catalog # 7767PE) l Adenosine 3’,5’cyclic monophosphate (cAMP) (Sigma, Catalog # A6885) 0.1 M stock in deionized water l Sialyltransferase Activity Kit (Catalog # EA002) l 96well Clear Plate (Costar, Catalog # 92592) l Plate Reader (Model: SpectraMax Plus by Molecular Devices) or equivalent Assay 1. Dilute 1 mM Phosphate Standard provided by the Sialyltransferase Kit by adding 40 µL of the 1 mM Phosphate Standard to 360 µL of Assay Buffer for a 100 µM stock. 2. Continue standard curve by performing six additional onehalf serial dilutions of the 100 µM Phosphate stock in Assay Buffer. -

Supporting Information
Supporting Information Koyanagi et al. 10.1073/pnas.0806215105 SI Materials and Methods CNGB, NM137763; human CNGB1, NM001297; human Amino Acid Sequences. The accession numbers of amino acid CNGB3, NM 019098; box jellyfish CNG, AB435552; fruit fly sequences used for analyses are as follows: Box jellyfish opsin, CNG4, NM 167441; fruit fly CNG3, NM 137871; human AB435549; sea anemone opsin, BR000662; hydra opsin, Con- CNGA4, NM 001037329; fruit fly CNGA, NM 057768; human CNGA1, NM000087; human CNGA2, NM005140; human tig39347:487820–488855 (http://hydrazome.metazome.net); hy- drozaon jellyfish opsin, AB332435; human encephalopsin, CNGA3, NM 001298. AF140242; ragworm c-opsin, AY692353; human rhodopsin, Western Blot Analysis. The proteins extracted from half a rhopalia U49742; human blue, M13299; human red, Z68193; lamprey were separated by 12% SDS/PAGE, transferred onto a PVDF parapinopsin, AB116380; lizard parietopsin, DQ100320; am- membrane, and incubated with 1:500 diluted anti-box jellyfish phioxus peropsin, AB050610; human peropsin, AF012270; hu- opsin antiserum. Visualization was carried out by the ABC man rgr, U15790; squid retinochrome, X57143; human melan- method (Vectastain) and by staining with 3,3Ј-diaminobenzidine opsin, AF147788; amphioxus melanopsin, AB205400; squid (Sigma). rhodopsin, X70498; fruit fly Rh1, K02315; human neuropsin, AY377391; amphioxus rhodopsin, AB050606; scallop Go- In Situ Hybridization. Digoxigenin-labeled antisense and sense RNA rhodopsin, AB006455; human muscarinic acetylcholine receptor probes for the box jellyfish opsin were synthesized by using the DIG M1 (CHRM1), NM000738; human melatonin receptor 1A RNA labeling kit (Roche). Sections were pretreated with protein- (MTNR1A), NM005958; Arabidopsis AKT1, U06745; Arabi- ase K and hybridized with each RNA probe. -

The G Protein-Coupled Receptor Subset of the Dog Genome Is More Similar
BMC Genomics BioMed Central Research article Open Access The G protein-coupled receptor subset of the dog genome is more similar to that in humans than rodents Tatjana Haitina1, Robert Fredriksson1, Steven M Foord2, Helgi B Schiöth*1 and David E Gloriam*2 Address: 1Department of Neuroscience, Functional Pharmacology, Uppsala University, BMC, Box 593, 751 24, Uppsala, Sweden and 2GlaxoSmithKline Pharmaceuticals, New Frontiers Science Park, 3rd Avenue, Harlow CM19 5AW, UK Email: Tatjana Haitina - [email protected]; Robert Fredriksson - [email protected]; Steven M Foord - [email protected]; Helgi B Schiöth* - [email protected]; David E Gloriam* - [email protected] * Corresponding authors Published: 15 January 2009 Received: 20 August 2008 Accepted: 15 January 2009 BMC Genomics 2009, 10:24 doi:10.1186/1471-2164-10-24 This article is available from: http://www.biomedcentral.com/1471-2164/10/24 © 2009 Haitina et al; licensee BioMed Central Ltd. This is an Open Access article distributed under the terms of the Creative Commons Attribution License (http://creativecommons.org/licenses/by/2.0), which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited. Abstract Background: The dog is an important model organism and it is considered to be closer to humans than rodents regarding metabolism and responses to drugs. The close relationship between humans and dogs over many centuries has lead to the diversity of the canine species, important genetic discoveries and an appreciation of the effects of old age in another species. The superfamily of G protein-coupled receptors (GPCRs) is one of the largest gene families in most mammals and the most exploited in terms of drug discovery. -

Wo 2010/075007 A2
(12) INTERNATIONAL APPLICATION PUBLISHED UNDER THE PATENT COOPERATION TREATY (PCT) (19) World Intellectual Property Organization International Bureau (10) International Publication Number (43) International Publication Date 1 July 2010 (01.07.2010) WO 2010/075007 A2 (51) International Patent Classification: (81) Designated States (unless otherwise indicated, for every C12Q 1/68 (2006.01) G06F 19/00 (2006.01) kind of national protection available): AE, AG, AL, AM, C12N 15/12 (2006.01) AO, AT, AU, AZ, BA, BB, BG, BH, BR, BW, BY, BZ, CA, CH, CL, CN, CO, CR, CU, CZ, DE, DK, DM, DO, (21) International Application Number: DZ, EC, EE, EG, ES, FI, GB, GD, GE, GH, GM, GT, PCT/US2009/067757 HN, HR, HU, ID, IL, IN, IS, JP, KE, KG, KM, KN, KP, (22) International Filing Date: KR, KZ, LA, LC, LK, LR, LS, LT, LU, LY, MA, MD, 11 December 2009 ( 11.12.2009) ME, MG, MK, MN, MW, MX, MY, MZ, NA, NG, NI, NO, NZ, OM, PE, PG, PH, PL, PT, RO, RS, RU, SC, SD, (25) Filing Language: English SE, SG, SK, SL, SM, ST, SV, SY, TJ, TM, TN, TR, TT, (26) Publication Language: English TZ, UA, UG, US, UZ, VC, VN, ZA, ZM, ZW. (30) Priority Data: (84) Designated States (unless otherwise indicated, for every 12/3 16,877 16 December 2008 (16.12.2008) US kind of regional protection available): ARIPO (BW, GH, GM, KE, LS, MW, MZ, NA, SD, SL, SZ, TZ, UG, ZM, (71) Applicant (for all designated States except US): DODDS, ZW), Eurasian (AM, AZ, BY, KG, KZ, MD, RU, TJ, W., Jean [US/US]; 938 Stanford Street, Santa Monica, TM), European (AT, BE, BG, CH, CY, CZ, DE, DK, EE, CA 90403 (US). -

From Inverse Agonism to 'Paradoxical Pharmacology' Richard A
International Congress Series 1249 (2003) 27-37 From inverse agonism to 'Paradoxical Pharmacology' Richard A. Bond*, Kenda L.J. Evans, Zsirzsanna Callaerts-Vegh Department of Pharmacological and Pharmaceutical Sciences, University of Houston, 521 Science and Research Bldg 2, 4800 Caltioun, Houston, TX 77204-5037, USA Received 16 April 2003; accepted 16 April 2003 Abstract The constitutive or spontaneous activity of G protein-coupled receptors (GPCRs) and compounds acting as inverse agonists is a recent but well-established phenomenon. Dozens of receptor subtypes for numerous neurotransmitters and hormones have been shown to posses this property. However, do to the apparently low percentage of receptors in the spontaneously active state, the physiologic relevance of these findings remains questionable. The possibility that the reciprocal nature of the effects of agonists and inverse agonists may extend to cellular signaling is discussed, and that this may account for the beneficial effects of certain p-adrenoceptor inverse agonists in the treatment of heart failure. © 2003 Elsevier Science B.V. All rights reserved. Keywords. Inverse agonism; GPCR; Paradoxical pharmacology 1. Brief history of inverse agonism at G protein-coupled receptors For approximately three-quarters of a century, ligands that interacted with G protein- coupled receptors (GPCRs) were classified either as agonists or antagonists. Receptors were thought to exist in a single quiescent state that could only induce cellular signaling upon agonist binding to the receptor to produce an activated state of the receptor. In this model, antagonists had no cellular signaling ability on their own, but did bind to the receptor and prevented agonists from being able to bind and activate the receptor. -
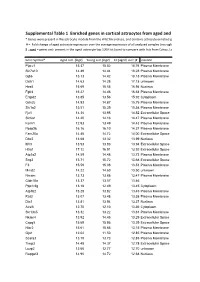
Supplemental Table 1 Enriched Genes in Cortical Astrocytes from Aged
Supplemental Table 1 Enriched genes in cortical astrocytes from aged and young-adult mice * Genes were present in the astrocyte module from the WGCNA analysis, and contains astrocyte enriched genes compared to microglia and oligodendrocytes # = Fold change of aged astrocyte expression over the average expression of all analyzed samples (microglia, astrocytes: young, old, with and without myelin contamination) $ ; aged = genes only present in the aged astrocyte top 1000 list (used to compare with lists from Cahoy, Lovatt, Doyle; see Fig. 4B), all = genes present in all astrocyte top 1000 lists Gene Symbol* Aged astr. (log2) Young astr.(log2) FC (aged/ aver.)# Location Ptprz1 15.37 15.02 18.76 Plasma Membrane Slc7a10 14.49 14.44 18.28 Plasma Membrane Gjb6 15.13 14.42 18.18 Plasma Membrane Dclk1 14.63 14.28 17.18 unknown Hes5 15.69 15.55 16.94 Nucleus Fgfr3 15.27 14.46 16.54 Plasma Membrane Entpd2 13.85 13.56 15.92 Cytoplasm Grin2c 14.93 14.87 15.75 Plasma Membrane Slc1a2 15.51 15.39 15.58 Plasma Membrane Fjx1 14.36 13.98 14.52 Extracellular Space Slc6a1 14.20 14.16 14.47 Plasma Membrane Kcnk1 12.93 13.49 14.43 Plasma Membrane Ppap2b 16.16 16.10 14.37 Plasma Membrane Fam20a 14.48 14.72 14.00 Extracellular Space Dbx2 13.68 13.32 13.99 Nucleus Itih3 13.93 13.93 13.94 Extracellular Space Htra1 17.12 16.91 13.92 Extracellular Space Atp1a2 14.59 14.48 13.73 Plasma Membrane Scg3 15.71 15.72 13.68 Extracellular Space F3 15.59 15.08 13.51 Plasma Membrane Mmd2 14.22 14.60 13.50 unknown Nrcam 13.73 13.88 13.47 Plasma Membrane Cldn10a 13.37 13.57 13.46 -

Genetic Basis of Idiopathic Scoliosis
Research & Review: Management of Cardiovascular and Orthopedic Complications Volume 1 Issue 1 Genetic Basis of Idiopathic Scoliosis S. Sreeremya Assistant Professor, Department of Biotechnology, Sree Narayana Guru College, Coimbatore, Tamil Nadu, India Email: [email protected] Abstract Idiopathic scoliosis (IS), the most usual spinal deformity, affects otherwise healthy children and adolescents during growth. The etiology is still not quiet understood, although genetic factors are believed to be important. This review corroborates the understanding of IS as a complex disease with a polygenic background. Presumably IS can be typically due to a spectrum of genetic risk variants, ranging from very rare or even private to very common. The most promising candidate genes are highlighted. Keywords: Idiopathic scoliosis, Genetics, Pathogenesis, Heredity INTRODUCTION marked by phenotypic complexity Idiopathic scoliosis (IS), the most general (variations in curve morphology and form of spinal deformity, affects otherwise magnitude, age of onset, rate of healthy children and adolescents during progression), and a prognosis mainly growth (Fig: 1). It usually presents as a rib ranging from increase in curve magnitude, hump visible at forward bending, together to stabilization, or to resolution with with unlevelled shoulders and an growth [5]. Genetic factors are known to asymmetrical waist [1]. According to play a pivotal role, as observed in twin Cobb, the diagnosis is specifically studies and their observation and singleton confirmed by a standing spinal radiograph multigenerational families [6]. A recent showing a lateral curvature of the spine research of monozygotic and dizygotic exceeding 10° [2]. A main concern in IS is twins from the Swedish twin registry the absence of reliable means by which to estimated that overall genetic effects predict risk of progression, leading to accounted for 39 % of the observed frequent follow-ups, radiographs, and phenotypic variance, leaving the remaining potentially unnecessary brace treatments. -

PDE4) Subtypes in Human Primary CD4+ T Cells: Predominant Role of PDE4D This Information Is Current As of September 26, 2021
Differential Expression and Function of Phosphodiesterase 4 (PDE4) Subtypes in Human Primary CD4+ T Cells: Predominant Role of PDE4D This information is current as of September 26, 2021. Daniel Peter, S. L. Catherine Jin, Marco Conti, Armin Hatzelmann and Christof Zitt J Immunol 2007; 178:4820-4831; ; doi: 10.4049/jimmunol.178.8.4820 http://www.jimmunol.org/content/178/8/4820 Downloaded from References This article cites 53 articles, 24 of which you can access for free at: http://www.jimmunol.org/content/178/8/4820.full#ref-list-1 http://www.jimmunol.org/ Why The JI? Submit online. • Rapid Reviews! 30 days* from submission to initial decision • No Triage! Every submission reviewed by practicing scientists • Fast Publication! 4 weeks from acceptance to publication by guest on September 26, 2021 *average Subscription Information about subscribing to The Journal of Immunology is online at: http://jimmunol.org/subscription Permissions Submit copyright permission requests at: http://www.aai.org/About/Publications/JI/copyright.html Email Alerts Receive free email-alerts when new articles cite this article. Sign up at: http://jimmunol.org/alerts The Journal of Immunology is published twice each month by The American Association of Immunologists, Inc., 1451 Rockville Pike, Suite 650, Rockville, MD 20852 Copyright © 2007 by The American Association of Immunologists All rights reserved. Print ISSN: 0022-1767 Online ISSN: 1550-6606. The Journal of Immunology Differential Expression and Function of Phosphodiesterase 4 :PDE4) Subtypes in Human Primary CD4؉ T Cells) Predominant Role of PDE4D1 Daniel Peter,* S. L. Catherine Jin,† Marco Conti,† Armin Hatzelmann,* and Christof Zitt2* Type 4 phosphodiesterases (PDE4) are critical regulators in TCR signaling by attenuating the negative constraint of cAMP. -

Table S1 the Four Gene Sets Derived from Gene Expression Profiles of Escs and Differentiated Cells
Table S1 The four gene sets derived from gene expression profiles of ESCs and differentiated cells Uniform High Uniform Low ES Up ES Down EntrezID GeneSymbol EntrezID GeneSymbol EntrezID GeneSymbol EntrezID GeneSymbol 269261 Rpl12 11354 Abpa 68239 Krt42 15132 Hbb-bh1 67891 Rpl4 11537 Cfd 26380 Esrrb 15126 Hba-x 55949 Eef1b2 11698 Ambn 73703 Dppa2 15111 Hand2 18148 Npm1 11730 Ang3 67374 Jam2 65255 Asb4 67427 Rps20 11731 Ang2 22702 Zfp42 17292 Mesp1 15481 Hspa8 11807 Apoa2 58865 Tdh 19737 Rgs5 100041686 LOC100041686 11814 Apoc3 26388 Ifi202b 225518 Prdm6 11983 Atpif1 11945 Atp4b 11614 Nr0b1 20378 Frzb 19241 Tmsb4x 12007 Azgp1 76815 Calcoco2 12767 Cxcr4 20116 Rps8 12044 Bcl2a1a 219132 D14Ertd668e 103889 Hoxb2 20103 Rps5 12047 Bcl2a1d 381411 Gm1967 17701 Msx1 14694 Gnb2l1 12049 Bcl2l10 20899 Stra8 23796 Aplnr 19941 Rpl26 12096 Bglap1 78625 1700061G19Rik 12627 Cfc1 12070 Ngfrap1 12097 Bglap2 21816 Tgm1 12622 Cer1 19989 Rpl7 12267 C3ar1 67405 Nts 21385 Tbx2 19896 Rpl10a 12279 C9 435337 EG435337 56720 Tdo2 20044 Rps14 12391 Cav3 545913 Zscan4d 16869 Lhx1 19175 Psmb6 12409 Cbr2 244448 Triml1 22253 Unc5c 22627 Ywhae 12477 Ctla4 69134 2200001I15Rik 14174 Fgf3 19951 Rpl32 12523 Cd84 66065 Hsd17b14 16542 Kdr 66152 1110020P15Rik 12524 Cd86 81879 Tcfcp2l1 15122 Hba-a1 66489 Rpl35 12640 Cga 17907 Mylpf 15414 Hoxb6 15519 Hsp90aa1 12642 Ch25h 26424 Nr5a2 210530 Leprel1 66483 Rpl36al 12655 Chi3l3 83560 Tex14 12338 Capn6 27370 Rps26 12796 Camp 17450 Morc1 20671 Sox17 66576 Uqcrh 12869 Cox8b 79455 Pdcl2 20613 Snai1 22154 Tubb5 12959 Cryba4 231821 Centa1 17897 -

Genetic Studies of Bipolar Disorder in Patients Selectedby Their Treatment
medigraphic Artemisaen línea Salud Mental 2008;31:431-440 Genetic studies of bipolar disorder Genetic studies of bipolar disorder in patients selected by their treatment response* Abigail Ortiz-Domínguez,1 Martin Alda1 Conferencia magistral SUMMARY rapid cycling and non-episodic course in the lamotrigine group) and co-morbidity, with the lamotrigine-responder group showing a higher Bipolar disorder (BD) is a major mood disorder with several genes frequency of panic attacks and substance abuse. of moderate or small effect contributing to the genetic susceptibility. In conclusion, pharmacogenetic studies may provide important It is also likely heterogeneous, which stimulated efforts to refine its clues to the nature of bipolar disorder and the response to long term clinical phenotype, studies investigating the link between BD treatment. susceptibility and response to a specific mood stabilizer appear to be one of the promising directions. Key words: Lithium response, pharmacogenetic, probands. In particular, excellent response to lithium prophylaxis has been described as a clinical marker of a more homogeneous subgroup of BD, characterized by an episodic course, low rates of co-morbid RESUMEN conditions, absence of rapid cycling, and a strong genetic loading. These results also suggest that lithium response clusters in families El trastorno bipolar (TB) es un trastorno afectivo con varios genes, de (independent of the increased familial loading for affective disorders), efecto leve o moderado, que contribuyen a su susceptibilidad. Es likely on a genetic basis. asimismo un trastorno heterogéneo, lo que ha estimulado diversas For almost 40 years, clinical studies have pointed to differences iniciativas para refinar el fenotipo de los pacientes con este trastor- between lithium responders (LR) and non-responders (LNR). -

Expression of Melatonin Receptors 1A/1B, Calmodulin and Estrogen Receptor 2 in Deep Paravertebral Muscles Revisited
MOLECULAR MEDICINE REPORTS 14: 5719-5724, 2016 Etiopathogenesis of adolescent idiopathic scoliosis: Expression of melatonin receptors 1A/1B, calmodulin and estrogen receptor 2 in deep paravertebral muscles revisited JOSEF ZAMECNIK1*, LENKA KRSKOVA1*, JAROMIR HACEK1, IVANA STETKAROVA2 and MARTIN KRBEC3 1Department of Pathology and Molecular Medicine, 2nd Faculty of Medicine, Charles University in Prague and University Hospital Motol, 15006 Prague; 2Department of Neurology, Charles University; 3Department of Orthopedics and Traumatology, 3rd Faculty of Medicine, Charles University in Prague and University Hospital Královské Vinohrady, 10034 Prague, Czech Republic Received October 28, 2015; Accepted October 11, 2016 DOI: 10.3892/mmr.2016.5927 Abstract. The pathogenesis of adolescent idiopathic scoliosis addition, no difference in expression was detected between the (AIS), including the associated local changes in deep para- patients with AIS and the controls. With regards to MTNR1A vertebral muscles, is poorly understood. The asymmetric and MTNR1B, their expression was very weak in paravertebral expression of several molecules involved in the melatonin muscles, and in the majority of cases their expression could signaling pathway, including melatonin receptors 1A/1B not be detected by repeated RT-qPCR analysis. Therefore, (MTNR1A/MTNR1B), estrogen receptor 2 (ESR2) and these data do not support the previously suggested role of the calmodulin (CALM1), has previously been suggested to be asymmetric expression of molecules involved in the melatonin associated with AIS. However, this hypothesis is based on signaling pathway in deep paravertebral muscles in the patho- single studies in which the data were obtained by different genesis of AIS. methodological approaches. Therefore, to evaluate the symmetry of the mRNA expression levels of these molecules, Introduction 18 patients with AIS and 10 non-scoliotic controls were enrolled in the present study. -

Noninvasive Sleep Monitoring in Large-Scale Screening of Knock-Out Mice
bioRxiv preprint doi: https://doi.org/10.1101/517680; this version posted January 11, 2019. The copyright holder for this preprint (which was not certified by peer review) is the author/funder, who has granted bioRxiv a license to display the preprint in perpetuity. It is made available under aCC-BY-ND 4.0 International license. Noninvasive sleep monitoring in large-scale screening of knock-out mice reveals novel sleep-related genes Shreyas S. Joshi1*, Mansi Sethi1*, Martin Striz1, Neil Cole2, James M. Denegre2, Jennifer Ryan2, Michael E. Lhamon3, Anuj Agarwal3, Steve Murray2, Robert E. Braun2, David W. Fardo4, Vivek Kumar2, Kevin D. Donohue3,5, Sridhar Sunderam6, Elissa J. Chesler2, Karen L. Svenson2, Bruce F. O'Hara1,3 1Dept. of Biology, University of Kentucky, Lexington, KY 40506, USA, 2The Jackson Laboratory, Bar Harbor, ME 04609, USA, 3Signal solutions, LLC, Lexington, KY 40503, USA, 4Dept. of Biostatistics, University of Kentucky, Lexington, KY 40536, USA, 5Dept. of Electrical and Computer Engineering, University of Kentucky, Lexington, KY 40506, USA. 6Dept. of Biomedical Engineering, University of Kentucky, Lexington, KY 40506, USA. *These authors contributed equally Address for correspondence and proofs: Shreyas S. Joshi, Ph.D. Dept. of Biology University of Kentucky 675 Rose Street 101 Morgan Building Lexington, KY 40506 U.S.A. Phone: (859) 257-2805 FAX: (859) 257-1717 Email: [email protected] Running title: Sleep changes in knockout mice bioRxiv preprint doi: https://doi.org/10.1101/517680; this version posted January 11, 2019. The copyright holder for this preprint (which was not certified by peer review) is the author/funder, who has granted bioRxiv a license to display the preprint in perpetuity.