The WD40 Repeat Protein Mutations
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

A Novel Breast Cancer ^ Associated BRIP1 (FANCJ/BACH1) Germ- Line Mutation Impairs Protein Stability and Function
Cancer Prevention and Susceptibility A Novel Breast Cancer ^ Associated BRIP1 (FANCJ/BACH1)Germ- line Mutation Impairs Protein Stability and Function Arcangela De Nicolo,1MariellaTancredi,4 Grazia Lombardi,4 Cristina Chantal Flemma,4 Serena Barbuti,4 Claudio Di Cristofano,4 Bijan Sobhian,1Generoso Bevilacqua,4 Ronny Drapkin,2,3 andMariaAdelaideCaligo4 Abstract Purpose: BRCA1-interacting protein 1 (BRIP1; FANCJ/BACH1), which encodes a DNA helicase that interacts with BRCA1, has been suggested to be a low-penetrance breast cancer predispos- ing gene.We aimed to assess whether BRIP1 mutations contribute to breast cancer susceptibility in our population and, if so, to investigate the effect of such mutation(s) on BRIP1function. Experimental Design: A series of49 breast/ovarian cancer families, devoid ofa BRCA1/ BRCA2 mutation, were screened for BRIP1 mutations. Functional analyses, including coimmuno- precipitation and stability assays, were employed to further characterize a previously unreported variant. Results: Five sequence alterations were identified, of which four had been already described. Herein, we report a novel BRIP1 germ-line mutation identified in a woman with early-onset breast cancer. The mutation consists ofa 4-nucleotide deletion (c.2992-2995delAAGA) in BRIP1 exon 20 that causes a shift in the reading frame, disrupts the BRCA1-binding domain of BRIP1, and creates a premature stop codon. Functional analysis ofthe recombinant mutant protein in transfected cells showed that the truncation interferes with the stability of the protein and with its ability to interact with BRCA1. Loss ofthe wild-type BRIP1 allele with retention ofthe mutated one was observed in the patient’s breast tumor tissue. Conclusions: These results, by showing that the newly identified BRIP1 c.2992-2995delAAGA mutation is associated with instability and functional impairment of the encoded protein, provide further evidence of a breast cancer ^ related role for BRIP1. -

Open Full Page
CCR PEDIATRIC ONCOLOGY SERIES CCR Pediatric Oncology Series Recommendations for Childhood Cancer Screening and Surveillance in DNA Repair Disorders Michael F. Walsh1, Vivian Y. Chang2, Wendy K. Kohlmann3, Hamish S. Scott4, Christopher Cunniff5, Franck Bourdeaut6, Jan J. Molenaar7, Christopher C. Porter8, John T. Sandlund9, Sharon E. Plon10, Lisa L. Wang10, and Sharon A. Savage11 Abstract DNA repair syndromes are heterogeneous disorders caused by around the world to discuss and develop cancer surveillance pathogenic variants in genes encoding proteins key in DNA guidelines for children with cancer-prone disorders. Herein, replication and/or the cellular response to DNA damage. The we focus on the more common of the rare DNA repair dis- majority of these syndromes are inherited in an autosomal- orders: ataxia telangiectasia, Bloom syndrome, Fanconi ane- recessive manner, but autosomal-dominant and X-linked reces- mia, dyskeratosis congenita, Nijmegen breakage syndrome, sive disorders also exist. The clinical features of patients with DNA Rothmund–Thomson syndrome, and Xeroderma pigmento- repair syndromes are highly varied and dependent on the under- sum. Dedicated syndrome registries and a combination of lying genetic cause. Notably, all patients have elevated risks of basic science and clinical research have led to important in- syndrome-associated cancers, and many of these cancers present sights into the underlying biology of these disorders. Given the in childhood. Although it is clear that the risk of cancer is rarity of these disorders, it is recommended that centralized increased, there are limited data defining the true incidence of centers of excellence be involved directly or through consulta- cancer and almost no evidence-based approaches to cancer tion in caring for patients with heritable DNA repair syn- surveillance in patients with DNA repair disorders. -

Analysis of and Fanconi Anemia Genes in -Negative Spanish Breast Cancer Families María J
Analysis of and Fanconi Anemia genes in -negative Spanish breast cancer families María J. García, Victoria Fernández, Ana Osorio, Alicia Barroso, Gemma Llort, Conxi Lázaro, Ignacio Blanco, Trinidad Caldés, Miguel Hoya, Teresa Ramón y Cajal, et al. To cite this version: María J. García, Victoria Fernández, Ana Osorio, Alicia Barroso, Gemma Llort, et al.. Analysis of and Fanconi Anemia genes in -negative Spanish breast cancer families. Breast Cancer Research and Treatment, Springer Verlag, 2008, 113 (3), pp.545-551. 10.1007/s10549-008-9945-0. hal-00478320 HAL Id: hal-00478320 https://hal.archives-ouvertes.fr/hal-00478320 Submitted on 30 Apr 2010 HAL is a multi-disciplinary open access L’archive ouverte pluridisciplinaire HAL, est archive for the deposit and dissemination of sci- destinée au dépôt et à la diffusion de documents entific research documents, whether they are pub- scientifiques de niveau recherche, publiés ou non, lished or not. The documents may come from émanant des établissements d’enseignement et de teaching and research institutions in France or recherche français ou étrangers, des laboratoires abroad, or from public or private research centers. publics ou privés. Breast Cancer Res Treat (2009) 113:545–551 DOI 10.1007/s10549-008-9945-0 EPIDEMIOLOGY Analysis of FANCB and FANCN/PALB2 Fanconi Anemia genes in BRCA1/2-negative Spanish breast cancer families Marı´a J. Garcı´a Æ Victoria Ferna´ndez Æ Ana Osorio Æ Alicia Barroso Æ Gemma LLort Æ Conxi La´zaro Æ Ignacio Blanco Æ Trinidad Calde´s Æ Miguel de la Hoya Æ Teresa Ramo´n y Cajal Æ Carmen Alonso Æ Marı´a-Isabel Tejada Æ Carlos San Roma´n Æ Luis Robles-Dı´az Æ Miguel Urioste Æ Javier Benı´tez Received: 12 February 2008 / Accepted: 12 February 2008 / Published online: 27 February 2008 Ó Springer Science+Business Media, LLC. -

The ATM Gene in Breast Cancer: Its Relevance in Clinical Practice
G C A T T A C G G C A T genes Review The ATM Gene in Breast Cancer: Its Relevance in Clinical Practice Luigia Stefania Stucci 1,* , Valeria Internò 1 , Marco Tucci 1,2 , Martina Perrone 1, Francesco Mannavola 1 , Raffaele Palmirotta 3 and Camillo Porta 1 1 Division of Medical Oncology, Department of Biomedical Sciences and Human Oncology, University of Bari ‘Aldo Moro’, A.O.U. Consorziale Policlinico di Bari, 70121 Bari, Italy; [email protected] (V.I.); [email protected] (M.T.); [email protected] (M.P.); [email protected] (F.M.); [email protected] (C.P.) 2 National Cancer Research Center, Tumori Institute IRCCS Giovanni Paolo II, 70121 Bari, Italy 3 Interdisciplinary Department of Medicine, Section of Sciences and Technologies of Laboratory Medicine, University of Bari, 70121 Bari, Italy; [email protected] * Correspondence: [email protected] Abstract: Molecular alterations of the Ataxia-telangiectasia (AT) gene are frequently detected in breast cancer (BC), with an incidence ranging up to 40%. The mutated form, the Ataxia-telangiectasia mutated (ATM) gene, is involved in cell cycle control, apoptosis, oxidative stress, and telomere maintenance, and its role as a risk factor for cancer development is well established. Recent studies have confirmed that some variants of ATM are associated with an increased risk of BC development and a worse prognosis. Thus, many patients harboring ATM mutations develop intermediate- and high-grade disease, and there is a higher rate of lymph node metastatic involvement. The evidence concerning a correlation of ATM gene mutations and the efficacy of therapeutic strategies in BC management are controversial. -

Ankrd9 Is a Metabolically-Controlled Regulator of Impdh2 Abundance and Macro-Assembly
ANKRD9 IS A METABOLICALLY-CONTROLLED REGULATOR OF IMPDH2 ABUNDANCE AND MACRO-ASSEMBLY by Dawn Hayward A dissertation submitted to The Johns Hopkins University in conformity with the requirements of the degree of Doctor of Philosophy Baltimore, Maryland April 2019 ABSTRACT Members of a large family of Ankyrin Repeat Domains proteins (ANKRD) regulate numerous cellular processes by binding and changing properties of specific protein targets. We show that interactions with a target protein and the functional outcomes can be markedly altered by cells’ metabolic state. ANKRD9 facilitates degradation of inosine monophosphate dehydrogenase 2 (IMPDH2), the rate-limiting enzyme in GTP biosynthesis. Under basal conditions ANKRD9 is largely segregated from the cytosolic IMPDH2 by binding to vesicles. Upon nutrient limitation, ANKRD9 loses association with vesicles and assembles with IMPDH2 into rod-like structures, in which IMPDH2 is stable. Inhibition of IMPDH2 with Ribavirin favors ANKRD9 binding to rods. The IMPDH2/ANKRD9 assembly is reversed by guanosine, which restores association of ANKRD9 with vesicles. The conserved Cys109Cys110 motif in ANKRD9 is required for the vesicles-to-rods transition as well as binding and regulation of IMPDH2. ANKRD9 knockdown increases IMPDH2 levels and prevents formation of IMPDH2 rods upon nutrient limitation. Thus, the status of guanosine pools affects the mode of ANKRD9 action towards IMPDH2. Advisor: Dr. Svetlana Lutsenko, Department of Physiology, Johns Hopkins University School of Medicine Second reader: -

About PALB2 Gene Mutations
About PALB2 Gene Mutations About Genes Recommendations Genes are in every cell in our bodies. Genes are made WOMEN of DNA, which gives instructions to cells about how to Starting at age 30: Mammogram and breast MRI every grow and work together. We have two copies of each year (scheduled 6 months apart) gene in each cell—one from our mother and one from our father. When genes work properly, they help stop Some medicines can lower the risk of getting breast cancer from developing. cancer. Surgery to remove both breasts may be an option for some women who have a strong family history of When it works right, the PALB2 gene works together breast cancer. with the BRCA1 and BRCA2 genes to help prevent WOMEN AND MEN cancer. Sometimes changes to the PALB2 gene happen. These changes are called mutations. Mutations can make Screening for pancreatic cancer has benefits and the PALB2 gene stop working and raise the risk for limitations. We do not recommend this screening for certain types of cancer. most people with PALB2 mutations. People who have a PALB2 mutation and a family history of pancreatic Having a mutation in the PALB2 gene makes your risk cancer should ask their doctor or genetic counselor for of getting breast and pancreatic cancers higher than more information. average. The risks for other cancers may also go up with KIDS AND SIBLINGS PALB2 mutations. Researchers are studying the PALB2 gene to understand more. Children and siblings of people with a PALB2 mutation have a 1 in 2 chance of also having the mutation. -

The Malignant Phenotype in Breast Cancer Is Driven by Eif4a1-Mediated Changes in the Translational Landscape
Citation: Cell Death and Disease (2015) 6, e1603; doi:10.1038/cddis.2014.542 OPEN & 2015 Macmillan Publishers Limited All rights reserved 2041-4889/15 www.nature.com/cddis The malignant phenotype in breast cancer is driven by eIF4A1-mediated changes in the translational landscape A Modelska1, E Turro1,2, R Russell1, J Beaton1, T Sbarrato3, K Spriggs4, J Miller1, S Gräf1,2,5, E Provenzano6,7, F Blows8, P Pharoah6,8, C Caldas1,6 and J Le Quesne*,1,3 Human mRNA DeXD/H-box helicases are ubiquitous molecular motors that are required for the majority of cellular processes that involve RNA metabolism. One of the most abundant is eIF4A, which is required during the initiation phase of protein synthesis to unwind regions of highly structured mRNA that would otherwise impede the scanning ribosome. Dysregulation of protein synthesis is associated with tumorigenesis, but little is known about the detailed relationships between RNA helicase function and the malignant phenotype in solid malignancies. Therefore, immunohistochemical analysis was performed on over 3000 breast tumors to investigate the relationship among expression of eIF4A1, the helicase-modulating proteins eIF4B, eIF4E and PDCD4, and clinical outcome. We found eIF4A1, eIF4B and eIF4E to be independent predictors of poor outcome in ER-negative disease, while in contrast, the eIF4A1 inhibitor PDCD4 was related to improved outcome in ER-positive breast cancer. Consistent with these data, modulation of eIF4A1, eIF4B and PCDC4 expression in cultured MCF7 cells all restricted breast cancer cell growth and cycling. The eIF4A1-dependent translatome of MCF7 cells was defined by polysome profiling, and was shown to be highly enriched for several classes of oncogenic genes, including G-protein constituents, cyclins and protein kinases, and for mRNAs with G/C-rich 5′UTRs with potential to form G-quadruplexes and with 3′UTRs containing microRNA target sites. -

A Novel LRRK2 Variant P.G2294R in the WD40 Domain Identified in Familial Parkinson's Disease Affects LRRK2 Protein Levels
International Journal of Molecular Sciences Article A Novel LRRK2 Variant p.G2294R in the WD40 Domain Identified in Familial Parkinson’s Disease Affects LRRK2 Protein Levels Jun Ogata 1, Kentaro Hirao 2, Kenya Nishioka 3 , Arisa Hayashida 3, Yuanzhe Li 3, Hiroyo Yoshino 4, Soichiro Shimizu 2, Nobutaka Hattori 1,3,4 and Yuzuru Imai 1,* 1 Department of Research for Parkinson’s Disease, Juntendo University Graduate School of Medicine, Tokyo 113-8421, Japan; [email protected] (J.O.); [email protected] (N.H.) 2 Department of Geriatric Medicine, Tokyo Medical University, 6-7-1 Nishishinjuku, Shinjuku-ku, Tokyo 160-0023, Japan; [email protected] (K.H.); [email protected] (S.S.) 3 Department of Neurology, Juntendo University School of Medicine, 2-1-1 Hongo, Bunkyo-ku, Tokyo 113-8421, Japan; [email protected] (K.N.); [email protected] (A.H.); [email protected] (Y.L.) 4 Research Institute for Diseases of Old Age, Graduate School of Medicine, Juntendo University, 2-1-1 Hongo, Bunkyo-ku, Tokyo 113-8421, Japan; [email protected] * Correspondence: [email protected]; Tel.: +81-3-6801-8332 Abstract: Leucine-rich repeat kinase 2 (LRRK2) is a major causative gene of late-onset familial Parkin- son’s disease (PD). The suppression of kinase activity is believed to confer neuroprotection, as most pathogenic variants of LRRK2 associated with PD exhibit increased kinase activity. We herein report a novel LRRK2 variant—p.G2294R—located in the WD40 domain, detected through targeted gene- Citation: Ogata, J.; Hirao, K.; panel screening in a patient with familial PD. -

Repeat Proteins Challenge the Concept of Structural Domains
844 Biochemical Society Transactions (2015) Volume 43, part 5 Repeat proteins challenge the concept of structural domains Rocıo´ Espada*1, R. Gonzalo Parra*1, Manfred J. Sippl†, Thierry Mora‡, Aleksandra M. Walczak§ and Diego U. Ferreiro*2 *Protein Physiology Lab, Dep de Qu´ımica Biologica, ´ Facultad de Ciencias Exactas y Naturales, UBA-CONICET-IQUIBICEN, Buenos Aires, C1430EGA, Argentina †Center of Applied Molecular Engineering, Division of Bioinformatics, Department of Molecular Biology, University of Salzburg, 5020 Salzburg, Austria ‡Laboratoire de physique statistique, CNRS, UPMC and Ecole normale superieure, ´ 24 rue Lhomond, 75005 Paris, France §Laboratoire de physique theorique, ´ CNRS, UPMC and Ecole normale superieure, ´ 24 rue Lhomond, 75005 Paris, France Abstract Structural domains are believed to be modules within proteins that can fold and function independently. Some proteins show tandem repetitions of apparent modular structure that do not fold independently, but rather co-operate in stabilizing structural forms that comprise several repeat-units. For many natural repeat-proteins, it has been shown that weak energetic links between repeats lead to the breakdown of co-operativity and the appearance of folding sub-domains within an apparently regular repeat array. The quasi-1D architecture of repeat-proteins is crucial in detailing how the local energetic balances can modulate the folding dynamics of these proteins, which can be related to the physiological behaviour of these ubiquitous biological systems. Introduction and between repeats, challenging the concept of structural It was early on noted that many natural proteins typically domain. collapse stretches of amino acid chains into compact units, defining structural domains [1]. These domains typically correlate with biological activities and many modern proteins can be described as composed by novel ‘domain arrange- Definition of the repeat-units ments’ [2]. -

A Structural Guide to Proteins of the NF-Kb Signaling Module
Downloaded from http://cshperspectives.cshlp.org/ on September 26, 2021 - Published by Cold Spring Harbor Laboratory Press A Structural Guide to Proteins of the NF-kB Signaling Module Tom Huxford1 and Gourisankar Ghosh2 1Department of Chemistry and Biochemistry, San Diego State University, 5500 Campanile Drive, San Diego, California 92182-1030 2Department of Chemistry and Biochemistry, University of California, San Diego, 9500 Gilman Drive, La Jolla, California 92093-0375 Correspondence: [email protected] The prosurvival transcription factor NF-kB specifically binds promoter DNA to activate target gene expression. NF-kB is regulated through interactions with IkB inhibitor proteins. Active proteolysis of these IkB proteins is, in turn, under the control of the IkB kinase complex (IKK). Together, these three molecules form the NF-kB signaling module. Studies aimed at charac- terizing the molecular mechanisms of NF-kB, IkB, and IKK in terms of their three-dimen- sional structures have lead to a greater understanding of this vital transcription factor system. F-kB is a master transcription factor that from the perspective of their three-dimensional Nresponds to diverse cell stimuli by activat- structures. ing the expression of stress response genes. Multiple signals, including cytokines, growth factors, engagement of the T-cell receptor, and NF-kB bacterial and viral products, induce NF-kB Introduction to NF-kB transcriptional activity (Hayden and Ghosh 2008). A point of convergence for the myriad NF-kB was discovered in the laboratory of of NF-kB inducing signals is the IkB kinase David Baltimore as a nuclear activity with bind- complex (IKK). Active IKK in turn controls ing specificity toward a ten-base-pair DNA transcription factor NF-kB by regulating pro- sequence 50-GGGACTTTCC-30 present within teolysis of the IkB inhibitor protein (Fig. -
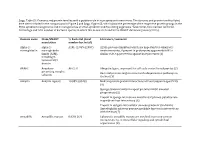
Supp. Table S2: Domains and Protein Families with a Putative Role in Host-Symbiont Interactions
Supp. Table S2: Domains and protein families with a putative role in host-symbiont interactions. The domains and protein families listed here were included in the comparisons in Figure 5 and Supp. Figure S5, which show the percentage of the respective protein groups in the Riftia symbiont metagenome and in metagenomes of other symbiotic and free-living organisms. % bacterial, total number bacterial: Percentage and total number of bacterial species in which this domain is found in the SMART database (January 2019). Domain name Pfam/SMART % bacterial (total Literature/comment annotation number bacterial) Alpha-2- alpha-2- A2M: 42.05% (2057) A2Ms: protease inhibitors which are important for eukaryotic macroglobulin macroglobulin innate immunity, if present in prokaryotes apparently fulfill a family (A2M), similar role, e.g. protection against host proteases (1) including N- terminal MG1 domain ANAPC Anaphase- APC2: 0 Ubiquitin ligase, important for cell cycle control in eukaryotes (2) promoting complex Bacterial proteins might interact with ubiquitination pathways in subunits the host (3) Ankyrin Ankyrin repeats 10.88% (8348) Mediate protein-protein interactions without sequence specificity (4) Sponge symbiont ankyrin-repeat proteins inhibit amoebal phagocytosis (5) Present in sponge microbiome metatranscriptomes, putative role in symbiont-host interactions (6) Present in obligate intracellular amoeba symbiont Candidatus Amoebophilus asiaticus genome, probable function in interactions with the host (7) Armadillo Armadillo repeats 0.83% (67) -

Distinct Roles of Different Fragments of PDCD4 in Regulating the Metastatic Behavior of B16 Melanoma Cells
INTERNATIONAL JOURNAL OF ONCOLOGY 42: 1725-1733, 2013 Distinct roles of different fragments of PDCD4 in regulating the metastatic behavior of B16 melanoma cells DI WANG, SHU GUO, SI-YUAN HAN, NAN XU, JIA-YAN GUO and QING SUN Department of Plastic Surgery, The First Hospital of China Medical University, Shenyang, Liaoning 110001, P.R. China Received December 12, 2012; Accepted January 29, 2013 DOI: 10.3892/ijo.2013.1841 Abstract. Melanoma is an aggressive cutaneous malignancy. In mice (7,8). As shown in Fig. 1, full length human PDCD4 this study, we demonstrated that the levels of the programmed contains an N-terminal putative RNA-binding domain cell death 4 (PDCD4) protein and mRNA were lower in tumor (residues 1-140) and two tandem MA3 domains, designated tissues compared with normal tissues. In order to further as nMA3 (residues 164-275) and cMA3 (residues 327-440) investigate the effects of PDCD4 and its fragments in B16 (9). The two MA3 domains have very similar structure and melanoma cells, we established B16 clones with expression of eIF4A-binding surfaces (10). The molecular basis for PDCD4- different PDCD4 fragments. Intact PDCD4, PDCD4∆164-469 mediated suppression has been linked to its high affinity and PDCD4∆327-440 expression, respectively, decreased MA-3 domains (11,12). NMR binding analysis has shown proliferation and migration and increased apoptosis in B16 that both MA3 domains of PDCD4 interacted with eIF4A cells in vitro. We found that intact PDCD4, PDCD4∆164-469 and prevented translation (10,11). However, another study has or PDCD4∆327-440 can inhibit the activity of MMP-2 and the demonstrated that the cMA3 domain alone is sufficient for expression of CXCR4.