(12) United States Patent (10) Patent No.: US 7,795,310 B2 Lee Et Al
Total Page:16
File Type:pdf, Size:1020Kb

Load more
Recommended publications
-

Nitrate Prodrugs Able to Release Nitric Oxide in a Controlled and Selective
Europäisches Patentamt *EP001336602A1* (19) European Patent Office Office européen des brevets (11) EP 1 336 602 A1 (12) EUROPEAN PATENT APPLICATION (43) Date of publication: (51) Int Cl.7: C07C 205/00, A61K 31/00 20.08.2003 Bulletin 2003/34 (21) Application number: 02425075.5 (22) Date of filing: 13.02.2002 (84) Designated Contracting States: (71) Applicant: Scaramuzzino, Giovanni AT BE CH CY DE DK ES FI FR GB GR IE IT LI LU 20052 Monza (Milano) (IT) MC NL PT SE TR Designated Extension States: (72) Inventor: Scaramuzzino, Giovanni AL LT LV MK RO SI 20052 Monza (Milano) (IT) (54) Nitrate prodrugs able to release nitric oxide in a controlled and selective way and their use for prevention and treatment of inflammatory, ischemic and proliferative diseases (57) New pharmaceutical compounds of general effects and for this reason they are useful for the prep- formula (I): F-(X)q where q is an integer from 1 to 5, pref- aration of medicines for prevention and treatment of in- erably 1; -F is chosen among drugs described in the text, flammatory, ischemic, degenerative and proliferative -X is chosen among 4 groups -M, -T, -V and -Y as de- diseases of musculoskeletal, tegumental, respiratory, scribed in the text. gastrointestinal, genito-urinary and central nervous sys- The compounds of general formula (I) are nitrate tems. prodrugs which can release nitric oxide in vivo in a con- trolled and selective way and without hypotensive side EP 1 336 602 A1 Printed by Jouve, 75001 PARIS (FR) EP 1 336 602 A1 Description [0001] The present invention relates to new nitrate prodrugs which can release nitric oxide in vivo in a controlled and selective way and without the side effects typical of nitrate vasodilators drugs. -
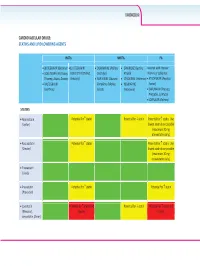
Statins and Lipid Lowering Agents
CARDIOVASCULAR Z/Ks^h>ZZh'^͗ ^dd/E^E>/W/>KtZ/E''Ed^ /E^d/Ɛ EEZd/Ɛ W/Ɛ x/d'Zs/Z ;ŝŬƚĂƌǀLJͿ x>s/d'Zs/Zͬ x KZs/Z/E ;WŝĨĞůƚƌŽ͕ x &s/ZE ;^ƵƐƚŝǀĂ͕ ŽŽƐƚĞĚǁŝƚŚƌŝƚŽŶĂǀŝƌ xK>hd'Zs/Z ;dŝǀŝĐĂLJ͕ K//^dd ;^ƚƌŝďŝůĚ͕ ĞůƐƚƌŝŐŽͿ ƚƌŝƉůĂͿ ;EŽƌǀŝƌͿŽƌĐŽďŝĐŝƐƚĂƚ dƌŝƵŵĞƋ͕:ƵůƵĐĂ͕ŽǀĂƚŽͿ 'ĞŶǀŽLJĂͿ x Z/>W/s/Z/E ;ĚƵƌĂŶƚ͕ x dZs/Z/E ;/ŶƚĞůĞŶĐĞͿ xdEs/Z ;ZĞLJĂƚĂnj͕ xZ>d'Zs/Z ŽŵƉůĞƌĂ͕KĚĞĨƐĞLJ͕ x Es/ZW/E ǀŽƚĂnjͿ ;/ƐĞŶƚƌĞƐƐͿ :ƵůƵĐĂͿ ;sŝƌĂŵƵŶĞͿ xZhEs/Z ;WƌĞnjŝƐƚĂ͕ WƌĞnjĐŽďŝdž͕^LJŵƚƵnjĂͿ x>KW/Es/Z ;<ĂůĞƚƌĂͿ ^dd/E^ xƚŽƌǀĂƐƚĂƚŝŶ WŽƚĞŶƚŝĂůĨŽƌ nƐƚĂƚŝŶ WŽƚĞŶƚŝĂůĨŽƌ pƐƚĂƚŝŶ WŽƚĞŶƚŝĂůĨŽƌ nƐƚĂƚŝŶ͘hƐĞ ;>ŝƉŝƚŽƌͿ ůŽǁĞƐƚƐƚĂƚŝŶĚŽƐĞƉŽƐƐŝďůĞ ;ŵĂdžŝŵƵŵϮϬŵŐ ĂƚŽƌǀĂƐƚĂƚŝŶĚĂŝůLJͿ͘ xZŽƐƵǀĂƐƚĂƚŝŶ WŽƚĞŶƚŝĂůĨŽƌ nƐƚĂƚŝŶ WŽƚĞŶƚŝĂůĨŽƌ nƐƚĂƚŝŶ͘hƐĞ ;ƌĞƐƚŽƌͿ ůŽǁĞƐƚƐƚĂƚŝŶĚŽƐĞƉŽƐƐŝďůĞ ;ŵĂdžŝŵƵŵϭϬŵŐ ƌŽƐƵǀĂƐƚĂƚŝŶĚĂŝůLJͿ͘ xWŝƚĂǀĂƐƚĂƚŝŶ ;>ŝǀĂůŽͿ xWƌĂǀĂƐƚĂƚŝŶ WŽƚĞŶƚŝĂůĨŽƌ nƐƚĂƚŝŶ WŽƚĞŶƚŝĂůĨŽƌ nƐƚĂƚŝŶ ;WƌĂǀĂĐŚŽůͿ x>ŽǀĂƐƚĂƚŝŶ WŽƚĞŶƚŝĂůĨŽƌ nƐƚĂƚŝŶĂŶĚ WŽƚĞŶƚŝĂůĨŽƌ pƐƚĂƚŝŶ WŽƚĞŶƚŝĂůĨŽƌ nƐƚĂƚŝŶĂŶĚ ;DĞǀĂĐŽƌͿ͕ ƚŽdžŝĐŝƚLJ ƚŽdžŝĐŝƚLJ ƐŝŵǀĂƐƚĂƚŝŶ ;ŽĐŽƌͿ CARDIOVASCULAR INSTIs NNRTIs PIs xBICTEGRAVIR (Biktarvy) xELVITEGRAVIR/ x DORAVIRINE (Pifeltro, x EFAVIRENZ (Sustiva, Boosted with ritonavir xDOLUTEGRAVIR (Tivicay, COBICISTAT (Stribild, Delstrigo) Atripla) (Norvir) or cobicistat Triumeq, Juluca) Genvoya) x RILPIVIRINE (Edurant, x ETRAVIRINE (Intelence) xATAZANAVIR (Reyataz, xRALTEGRAVIR Complera, Odefsey, x NEVIRAPINE Evotaz) (Isentress) Juluca) (Viramune) xDARUNAVIR (Prezista, Prezcobix, Symtuza) xLOPINAVIR (Kaletra) FIBRATES xFenofibrate, bezafibrate, gemfibrozil CHOLESTEROL ABSORPTION INHIBITOR xEzetimibe -

Genetically Modified Baculoviruses for Pest
INSECT CONTROL BIOLOGICAL AND SYNTHETIC AGENTS This page intentionally left blank INSECT CONTROL BIOLOGICAL AND SYNTHETIC AGENTS EDITED BY LAWRENCE I. GILBERT SARJEET S. GILL Amsterdam • Boston • Heidelberg • London • New York • Oxford Paris • San Diego • San Francisco • Singapore • Sydney • Tokyo Academic Press is an imprint of Elsevier Academic Press, 32 Jamestown Road, London, NW1 7BU, UK 30 Corporate Drive, Suite 400, Burlington, MA 01803, USA 525 B Street, Suite 1800, San Diego, CA 92101-4495, USA ª 2010 Elsevier B.V. All rights reserved The chapters first appeared in Comprehensive Molecular Insect Science, edited by Lawrence I. Gilbert, Kostas Iatrou, and Sarjeet S. Gill (Elsevier, B.V. 2005). All rights reserved. No part of this publication may be reproduced or transmitted in any form or by any means, electronic or mechanical, including photocopy, recording, or any information storage and retrieval system, without permission in writing from the publishers. Permissions may be sought directly from Elsevier’s Rights Department in Oxford, UK: phone (þ44) 1865 843830, fax (þ44) 1865 853333, e-mail [email protected]. Requests may also be completed on-line via the homepage (http://www.elsevier.com/locate/permissions). Library of Congress Cataloging-in-Publication Data Insect control : biological and synthetic agents / editors-in-chief: Lawrence I. Gilbert, Sarjeet S. Gill. – 1st ed. p. cm. Includes bibliographical references and index. ISBN 978-0-12-381449-4 (alk. paper) 1. Insect pests–Control. 2. Insecticides. I. Gilbert, Lawrence I. (Lawrence Irwin), 1929- II. Gill, Sarjeet S. SB931.I42 2010 632’.7–dc22 2010010547 A catalogue record for this book is available from the British Library ISBN 978-0-12-381449-4 Cover Images: (Top Left) Important pest insect targeted by neonicotinoid insecticides: Sweet-potato whitefly, Bemisia tabaci; (Top Right) Control (bottom) and tebufenozide intoxicated by ingestion (top) larvae of the white tussock moth, from Chapter 4; (Bottom) Mode of action of Cry1A toxins, from Addendum A7. -

Investigations Into the Role of Endothelin, Nitric Oxide And
INVESTIGATIONS INTO THE ROLE OF ENDOTHELIN, NITRIC OXIDE AND PROSTAGLANDINS IN THE PATHOGENESIS OF DIABETIC CYSTOPATHY. A thesis presentedfor the degree o f Doctor o f Medicine in the Faculty o f Medicine of the University o f London Submitted By FAIZ HASSAN MUMTAZ, FRCS. Departments of Urology, Molecular Pathology and Clinical Biochemistry and Academic Surgery Royal Free and University College Medical School (University College London) and The Royal Free Hampstead NHS Trust, Pond Street London NW3 2QG ProQuest Number: 10014733 All rights reserved INFORMATION TO ALL USERS The quality of this reproduction is dependent upon the quality of the copy submitted. In the unlikely event that the author did not send a complete manuscript and there are missing pages, these will be noted. Also, if material had to be removed, a note will indicate the deletion. uest. ProQuest 10014733 Published by ProQuest LLC(2016). Copyright of the Dissertation is held by the Author. All rights reserved. This work is protected against unauthorized copying under Title 17, United States Code. Microform Edition © ProQuest LLC. ProQuest LLC 789 East Eisenhower Parkway P.O. Box 1346 Ann Arbor, Ml 48106-1346 ABSTRACT Endothelin-1 (ET- 1) and its receptors (ETa and ETb) have been identified on the urothelium and smooth muscle of the urinary bladder. ET-1 has potent smooth muscle contractile and mitogenic properties. In contrast, the nitric oxide (NO)-cyclic-guanine 3’5’ monophosphate (cGMP) pathway mediates bladder outlet smooth muscle relaxation. In addition, the prostaglandin (PG)-cyclic-adenosine 3’5’ monophosphate (cAMP) pathway regulates urinary tract smooth muscle tone. -

Classification Decisions Taken by the Harmonized System Committee from the 47Th to 60Th Sessions (2011
CLASSIFICATION DECISIONS TAKEN BY THE HARMONIZED SYSTEM COMMITTEE FROM THE 47TH TO 60TH SESSIONS (2011 - 2018) WORLD CUSTOMS ORGANIZATION Rue du Marché 30 B-1210 Brussels Belgium November 2011 Copyright © 2011 World Customs Organization. All rights reserved. Requests and inquiries concerning translation, reproduction and adaptation rights should be addressed to [email protected]. D/2011/0448/25 The following list contains the classification decisions (other than those subject to a reservation) taken by the Harmonized System Committee ( 47th Session – March 2011) on specific products, together with their related Harmonized System code numbers and, in certain cases, the classification rationale. Advice Parties seeking to import or export merchandise covered by a decision are advised to verify the implementation of the decision by the importing or exporting country, as the case may be. HS codes Classification No Product description Classification considered rationale 1. Preparation, in the form of a powder, consisting of 92 % sugar, 6 % 2106.90 GRIs 1 and 6 black currant powder, anticaking agent, citric acid and black currant flavouring, put up for retail sale in 32-gram sachets, intended to be consumed as a beverage after mixing with hot water. 2. Vanutide cridificar (INN List 100). 3002.20 3. Certain INN products. Chapters 28, 29 (See “INN List 101” at the end of this publication.) and 30 4. Certain INN products. Chapters 13, 29 (See “INN List 102” at the end of this publication.) and 30 5. Certain INN products. Chapters 28, 29, (See “INN List 103” at the end of this publication.) 30, 35 and 39 6. Re-classification of INN products. -

Enzyme Mediated Inflammation: Epidemiology and Therapeutic Implications Parteek Prasher*
Available online www.jocpr.com Journal of Chemical and Pharmaceutical Research, 2016, 8(9):89-100 ISSN : 0975-7384 Review Article CODEN(USA) : JCPRC5 Enzyme Mediated Inflammation: Epidemiology and Therapeutic implications Parteek Prasher* Assistant Professor of Chemistry, University of Petroleum and Energy Study, Energy acres, Uttrakhand, India ___________________________________________________________________________________ ABSTRACT The identification of various enzymes participating in the arachidonic acid metabolism and recognition of inflammatory metabolites led to systematic studies for the development of anti-inflammatory drugs. Starting with the stimulatory role of phospholipases to release arachidonic acid from phospholipids, over-expression of cyclooxygenase-2 (COX-2) and lipoxygenase (LOX) enzymes cause more than normal production of the respective metabolites. Excessive production of prostaglandins and leukotrienes leads to the manifestation of acute to chronic inflammation in humans. The consequent ailments are expressed in the form of asthma, atherosclerosis, irritable bowel syndrome, arthritis and cancer. Targeting the induced isoform of cyclooxygenase viz. cyclooxygenase-2 (COX-2) and 5-LOX enzyme has opened new aspects for capping the enzyme mediated inflammation. A number of such aspects have been discussed in this comprehensive review. Keywords: COX; Cyclooxygenase; LOX; Lipoxygenase; Arachidonic acid ___________________________________________________________________________________ INTRODUCTION According to a recent survey of the World Health Organization (WHO), cancer causes around 13% of the total deaths worldwide and if it continues rising, it is estimated to cause 13.1 million deaths in 2030 [1]. Moreover, the reason for about 15% of the total cancer deaths has been reported [2] to be associated with chronic inflammation. Hence, an obvious way out to minimize cancer deaths is to put control over the inflammatory conditions. -

Clinofibrate Improved Canine Lipid Metabolism in Some but Not All Breeds
NOTE Internal Medicine Clinofibrate improved canine lipid metabolism in some but not all breeds Yohtaro SATO1), Nobuaki ARAI2), Hidemi YASUDA3) and Yasushi MIZOGUCHI4)* 1)Graduate School of Agriculture, Meiji University, 1-1-1 Higashimita, Tama-ku, Kawasaki, Kanagawa 214-8571, Japan 2)Spectrum Lab Japan, 1-5-22-201 Midorigaoka, Meguro-ku, Tokyo 152-0034, Japan 3)Yasuda Veterinary Clinic, 1-5-22 Midorigaoka, Meguro-ku, Tokyo 152-0034, Japan 4)School of Agriculture, Meiji University, 1-1-1 Higashimita, Tama-ku, Kawasaki, Kanagawa 214-8571, Japan ABSTRACT. The objectives of this study were to assess if Clinofibrate (CF) treatment improved J. Vet. Med. Sci. lipid metabolism in dogs, and to clarify whether its efficacy is influenced by canine characteristics. 80(6): 945–949, 2018 We collected medical records of 306 dogs and performed epidemiological analyses. Lipid values of all lipoproteins were significantly decreased by CF medication, especially VLDL triglyceride doi: 10.1292/jvms.17-0703 (TG) concentration (mean reduction rate=54.82%). However, 17.65% of dogs showed drug refractoriness in relation to TG level, and Toy Poodles had a lower CF response than other breeds (OR=5.36, 95% CI=2.07–13.90). Therefore, our study suggests that genetic factors may have an Received: 22 December 2017 effect on CF response, so genetic studies on lipid metabolism-related genes might be conducted Accepted: 9 March 2018 to identify variations in CF efficacy. Published online in J-STAGE: KEY WORDS: clinofibrate, descriptive epidemiology, drug response, dyslipidemia, Toy Poodle 26 March 2018 High serum cholesterol (Cho) and triglyceride (TG) concentrations in dogs are caused by various factors such as lack of exercise, high fat diets, obesity, neutralization, age, diseases and breed [6, 21, 24]. -

(12) United States Patent (10) Patent No.: US 6,264,917 B1 Klaveness Et Al
USOO6264,917B1 (12) United States Patent (10) Patent No.: US 6,264,917 B1 Klaveness et al. (45) Date of Patent: Jul. 24, 2001 (54) TARGETED ULTRASOUND CONTRAST 5,733,572 3/1998 Unger et al.. AGENTS 5,780,010 7/1998 Lanza et al. 5,846,517 12/1998 Unger .................................. 424/9.52 (75) Inventors: Jo Klaveness; Pál Rongved; Dagfinn 5,849,727 12/1998 Porter et al. ......................... 514/156 Lovhaug, all of Oslo (NO) 5,910,300 6/1999 Tournier et al. .................... 424/9.34 FOREIGN PATENT DOCUMENTS (73) Assignee: Nycomed Imaging AS, Oslo (NO) 2 145 SOS 4/1994 (CA). (*) Notice: Subject to any disclaimer, the term of this 19 626 530 1/1998 (DE). patent is extended or adjusted under 35 O 727 225 8/1996 (EP). U.S.C. 154(b) by 0 days. WO91/15244 10/1991 (WO). WO 93/20802 10/1993 (WO). WO 94/07539 4/1994 (WO). (21) Appl. No.: 08/958,993 WO 94/28873 12/1994 (WO). WO 94/28874 12/1994 (WO). (22) Filed: Oct. 28, 1997 WO95/03356 2/1995 (WO). WO95/03357 2/1995 (WO). Related U.S. Application Data WO95/07072 3/1995 (WO). (60) Provisional application No. 60/049.264, filed on Jun. 7, WO95/15118 6/1995 (WO). 1997, provisional application No. 60/049,265, filed on Jun. WO 96/39149 12/1996 (WO). 7, 1997, and provisional application No. 60/049.268, filed WO 96/40277 12/1996 (WO). on Jun. 7, 1997. WO 96/40285 12/1996 (WO). (30) Foreign Application Priority Data WO 96/41647 12/1996 (WO). -

Pharmacological Intervention in Preterm Labour
PDF hosted at the Radboud Repository of the Radboud University Nijmegen The following full text is a publisher's version. For additional information about this publication click this link. http://hdl.handle.net/2066/146284 Please be advised that this information was generated on 2021-10-07 and may be subject to change. 4 --. 'HARMACOLLOÒICAUNTERVENTIO N IN ?RESEW\KA]ÍÓ[JT PHARMACOLOGICAL INTERVENTION IN PRETERM LABOUR Dose studies on ritodrine administration C.A.G. HOLLEBOOM Druk: Haveka bv, Alblasserdam I.S.B.N. 90-9009763-5 Thesis Catholic University Nijmegen.- With réf.- With summary in dutch Subject heading: preterm labour/ tocolysis/ ritodrine Financial support for the publication of this thesis by Solvay Pharma bv, Research Fonds Bronovo, Novo Nordisk Farma bv, Schering Nederland bv, Organon Nederland bv, Glaxo Wellcome bv, Ferring bv, Pfizer bv, Wyeth Laboratoria bv and Zeneca bv, is gratefully acknowledged. Cover illustration: Roses; flowers of love, life and birth.* Cover design: Annemieke van Rosmalen * J. Hall. Dictionary of subjects and symbols in art. New York, 1979. PHARMACOLOGICAL INTERVENTION IN PRETERM LABOUR Dose studies on ritodrine administration een wetenschappelijke proeve op het gebied van de Medische Wetenschappen Proefschrift ter verkrijging van de graad van doctor aan de Katholieke Universiteit Nijmegen, volgens besluit van het College van Decanen in het openbaar te verdedigen op dinsdag 26 november 1996 des namiddags om '3.30 uur precies door Caspar Adriaan Godfried Holleboom geboren op 30 april 1955 te Lubuk Pakan, Indonesië Promotores: Prof. Dr. J.M.W.M. Merkus Prof. Dr. M.J.N.C. Keirse, Flinders University, Adelaide, Australia Manuscriptcommissie: Prof. -

Effects of Clofibrate Derivatives on Hyperlipidemia Induced by a Cholesterol-Free, High-Fructose Diet in Rats
Showa Univ. J. Med. Sci. 7(2), 173•`182, December 1995 Original Effects of Clofibrate Derivatives on Hyperlipidemia Induced by a Cholesterol-Free, High-Fructose Diet in Rats Hideyukl KURISHIMA,Sadao NAKAYAMA,Minoru FURUYA and Katsuji OGUCHI Abstract: The effects of the clofibrate derivatives fenofibrate (FF), bezafibrate (BF), and clinofibrate (CF), on hyperlipidemia induced by a cholesterol-free, high-fructose diet (HFD) in rats were investigated. Feeding of HFD for 2 weeks increased the high-density lipoprotein subfraction (HDL1) and decreased the low-density lipoprotein (LDL) fraction. The levels of total cholesterol (TC), free cholesterol, triglyceride (TG), and phospholipid in serum were increased by HFD feeding. Administration of CF inhibited the increase in HDL1 content. All three agents inhibited the decrease in LDL level. Both BF and CF decreased VLDL level. Administration of FF, BF, or CF inhibited the increases of serum lipids, especially that of TC and TG. The inhibitory effects of CF on HFD- induced increases in HDL1, TC, and TG were greater than those of FF and BF. These results demonstrate that FF, BF, and CF improve the intrinsic hyper- lipidemia induced by HFD feeding in rats. Key words: fenofibrate, bezafibrate, clinofibrate, fructose-induced hyperlipide- mia, lipoprotein. Introduction Clofibrate is one of the most effective antihypertriglycedemic agents currently available. However, because of its adverse effects, such as hepatomegaly1, several derivatives, such as clinofibrate (CF) and bezafibrate (BF) have been developed which are more effective and have fewer adverse effects. For example, it has been shown that the hypolipidemic effect of CF is greater than that of clofibrate while its tendency to produce hepatomegaly is less1. -

Toiminta Unita on Ulla La Mungukurti |
TOIMINTAUNITA USON 20180071390A1ULLA LA MUNGUKURTI | ( 19) United States (12 ) Patent Application Publication (10 ) Pub. No. : US 2018/ 0071390 A1 PATEL et al. (43 ) Pub . Date : Mar . 15 , 2018 ( 54 ) COMPOSITIONS OF PHARMACEUTICAL A61K 9 / 06 (2006 .01 ) ACTIVES CONTAINING DIETHYLENE A61K 9 /00 (2006 .01 ) GLYCOL MONOETHYL ETHER OR OTHER A61K 31 /573 ( 2006 .01 ) ALKYL DERIVATIVES A61K 31/ 565 ( 2006 .01 ) A61K 31/ 4439 ( 2006 . 01 ) ( 71 ) Applicant : THEMIS MEDICARE LIMITED , A61K 31 / 167 ( 2006 . 01 ) Mumbai (IN ) A61K 31 / 57 (2006 . 01) (52 ) U . S . CI. (72 ) Inventors : Dinesh Shantilal PATEL , Mumbai CPC .. .. .. A61K 47 / 10 ( 2013 . 01 ) ; A61K 9 /4858 ( IN ) ; Sachin Dinesh PATEL , Mumbai ( 2013 .01 ) ; A61K 9 /08 ( 2013 .01 ) ; A61K 9 / 06 ( IN ) ; Shashikant Prabhudas ( 2013 .01 ) ; A61K 9 / 0014 ( 2013 .01 ) ; A61K KURANI, Mumbai ( IN ) ; Madhavlal 31/ 573 ( 2013 .01 ) ; A61K 31 /57 ( 2013 .01 ) ; Govindlal PATEL , Mumbai ( IN ) A61K 31/ 565 ( 2013 .01 ) ; A61K 31 /4439 (73 ) Assignee : THEMIS MEDICARE LIMITED , ( 2013 .01 ) ; A61K 31/ 167 ( 2013 .01 ) ; A61K Mumbai (IN ) 9 /0048 ( 2013 .01 ) ; A61K 9 /0019 (2013 .01 ) ( 57 ) ABSTRACT (21 ) Appl. No .: 15 / 801, 390 The present invention relates to pharmaceutical composi tions of various pharmaceutical actives, especially lyophilic ( 22 ) Filed : Nov . 2 , 2017 and hydrophilic actives containing Diethylene glycol mono ethyl ether or other alkyl derivatives thereof as a primary Related U . S . Application Data vehicle and /or to pharmaceutical compositions utilizing (62 ) Division of application No. 14 /242 , 973 , filed on Apr. Diethylene glycol monoethyl ether or other alkyl derivatives 2 , 2014 , now Pat. No. 9 , 827 ,315 . -

Actions of Non-Steroidal Anti-Inflammatory Drugs in Sheep
ACTIONS OF NON-STEROIDAL ANTI-INFLAMMATORY DRUGS IN SHEEP by Elizabeth McCaulI Welsh B.V.M.S. , M.R.C.V.S. A thesis submitted for the degree of Doctor of Philosophy in the Faculty of Veterinary Medicine of the University of Glasgow Department of Veterinary Pharmacology August, 1993. ProQuest Number: 11007781 All rights reserved INFORMATION TO ALL USERS The quality of this reproduction is dependent upon the quality of the copy submitted. In the unlikely event that the author did not send a com plete manuscript and there are missing pages, these will be noted. Also, if material had to be removed, a note will indicate the deletion. uest ProQuest 11007781 Published by ProQuest LLC(2018). Copyright of the Dissertation is held by the Author. All rights reserved. This work is protected against unauthorized copying under Title 17, United States C ode Microform Edition © ProQuest LLC. ProQuest LLC. 789 East Eisenhower Parkway P.O. Box 1346 Ann Arbor, Ml 48106- 1346 GLASGOW UNIVERSITY LIBRARY i To my parents and Chris. CONTENTS Acknowledgements iv Declaration v Summary vi List of figures ix List of tables xii Abbreviations xvii Chapter 1- General Introduction 1 Chapter 2- General Materials and Methods 24 Chapter 3- The antinociceptive effects of non-steroidal anti-inflammatory drugs in sheep: 1 Clinical pain 36 3 .1 -Introduction 37 3.2- Materials and methods 47 3.3- Results 53 3.4- Discussion 98 Chapter 4- The antinociceptive effects of non-steroidal anti-inflammatory drugs in sheep: 2 Experimental pain 112 4.1-Introduction 113 4.2- Materials and methods 120 4.3- Results 123 4.4- Discussion 141 Chapter 5- The Pharmacokinetics of flunixin meglumine and carprofen (racemate) 150 iii 5.1- Introduction 151 5.2- Materials and methods 154 5.3- Results 159 5.4- Discussion 173 Chapter 6- Methods of lameness measurement 179 6.1- Introduction 180 6.2- Materials and methods 187 6.3- Results 192 6.4- Discussion 209 Chapter 7- General Discussion 218 References 226 ACKNOWLEDGEMENTS This work was generously funded by the Agricultural and Food Research Council.